

Genome-wide quantification of transcription factor binding at single-DNA-molecule resolution using methyl-transferase footprinting
- PMID: 34773120
- PMCID: PMC7613001
- DOI: 10.1038/s41596-021-00630-1
Genome-wide quantification of transcription factor binding at single-DNA-molecule resolution using methyl-transferase footprinting
Abstract
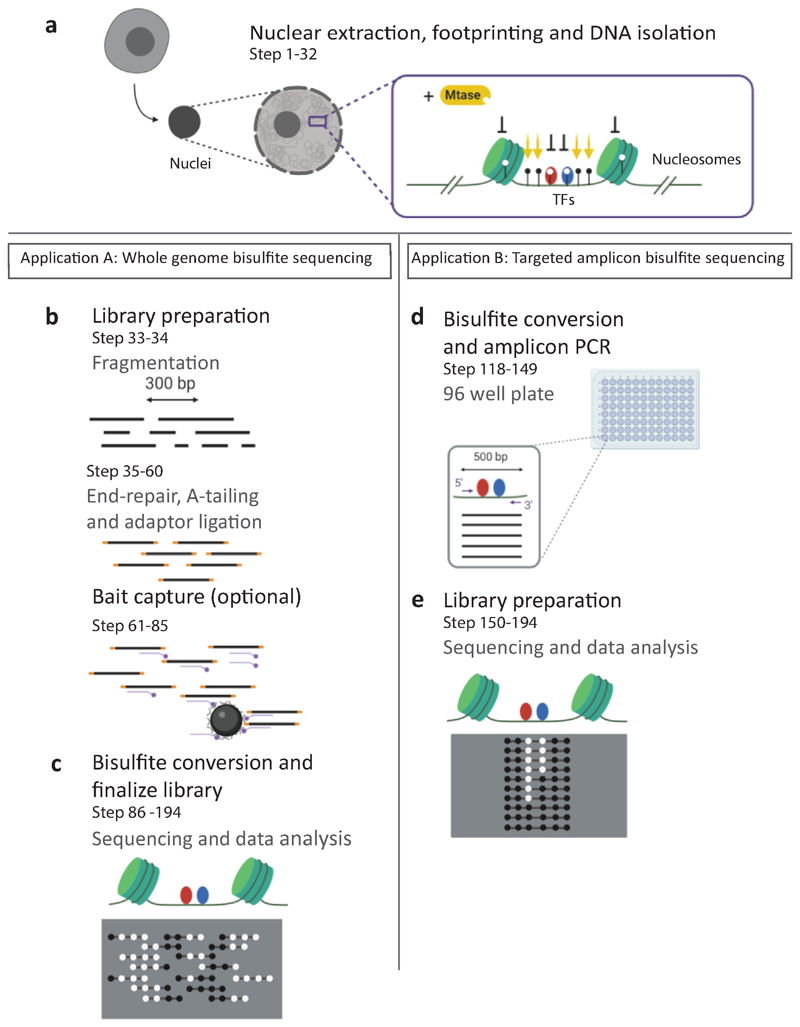
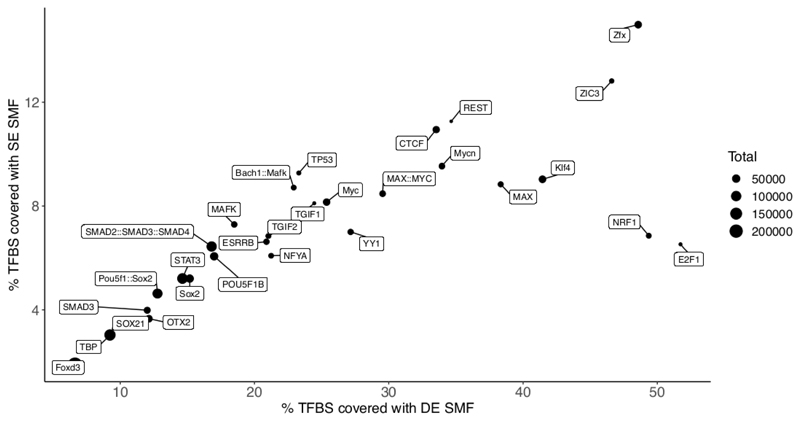
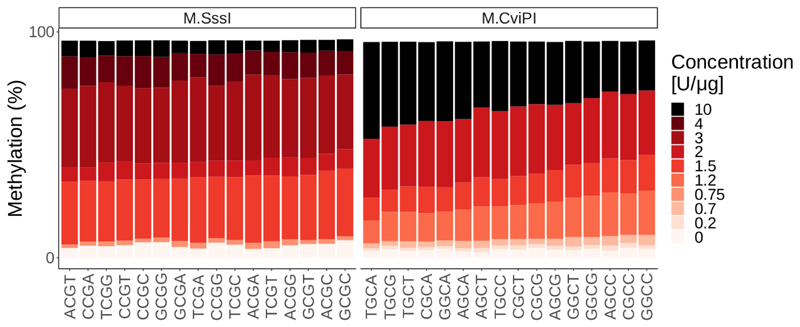
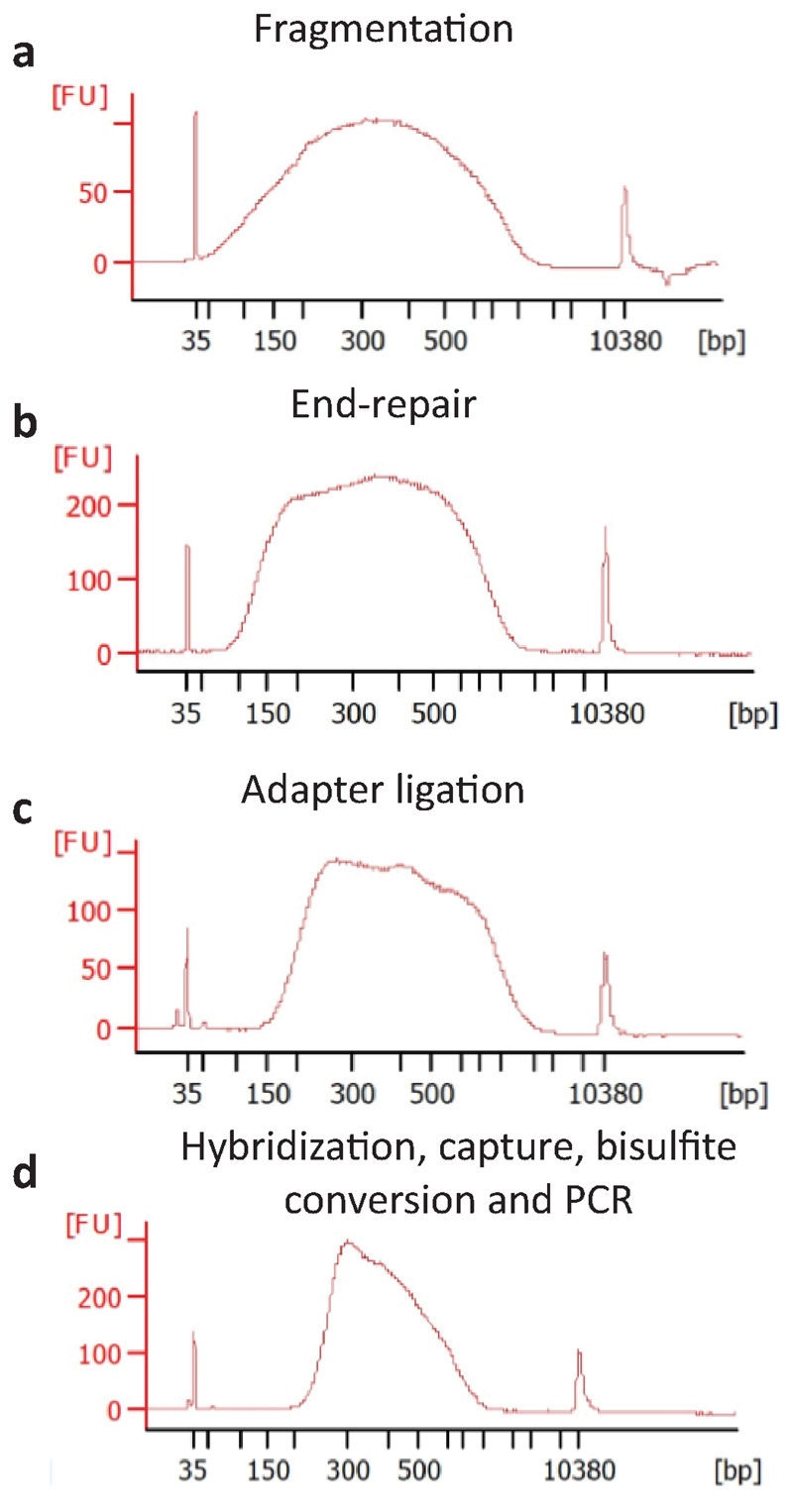
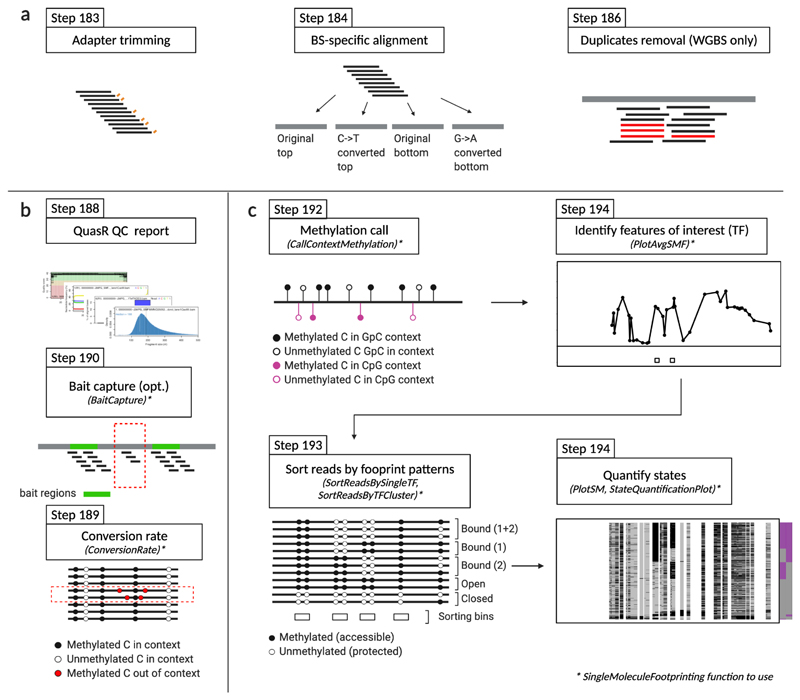
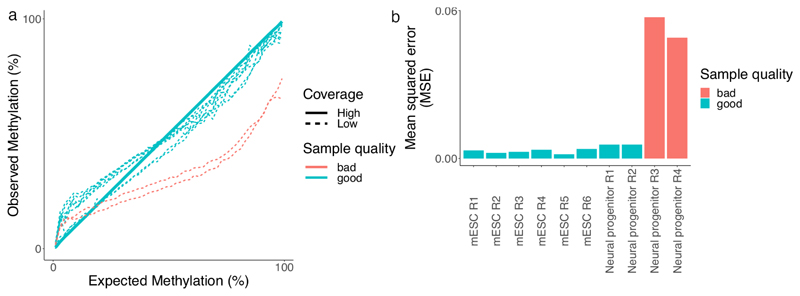
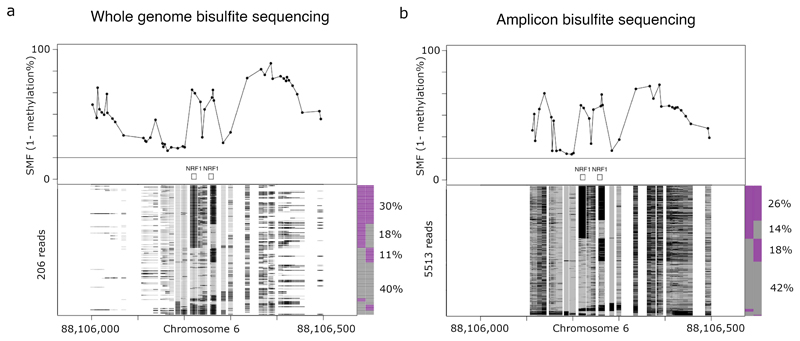
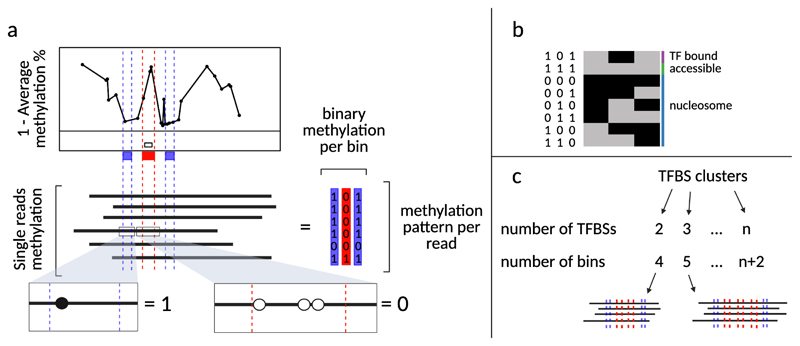
Precise control of gene expression requires the coordinated action of multiple factors at cis-regulatory elements. We recently developed single-molecule footprinting to simultaneously resolve the occupancy of multiple proteins including transcription factors, RNA polymerase II and nucleosomes on single DNA molecules genome-wide. The technique combines the use of cytosine methyltransferases to footprint the genome with bisulfite sequencing to resolve transcription factor binding patterns at cis-regulatory elements. DNA footprinting is performed by incubating permeabilized nuclei with recombinant methyltransferases. Upon DNA extraction, whole-genome or targeted bisulfite libraries are prepared and loaded on Illumina sequencers. The protocol can be completed in 4-5 d in any laboratory with access to high-throughput sequencing. Analysis can be performed in 2 d using a dedicated R package and requires access to a high-performance computing system. Our method can be used to analyze how transcription factors cooperate and antagonize to regulate transcription.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Molecular Co-occupancy Identifies Transcription Factor Binding Cooperativity In Vivo.Mol Cell. 2021 Jan 21;81(2):255-267.e6. doi: 10.1016/j.molcel.2020.11.015. Epub 2020 Dec 7. Mol Cell. 2021. PMID: 33290745 Free PMC article.
-
Constitutive nucleosome depletion and ordered factor assembly at the GRP78 promoter revealed by single molecule footprinting.PLoS Genet. 2006 Sep 22;2(9):e160. doi: 10.1371/journal.pgen.0020160. PLoS Genet. 2006. PMID: 17002502 Free PMC article.
-
Massively multiplex single-molecule oligonucleosome footprinting.Elife. 2020 Dec 2;9:e59404. doi: 10.7554/eLife.59404. Elife. 2020. PMID: 33263279 Free PMC article.
-
Studying transcription factor function in the genome at molecular resolution.Trends Genet. 2021 Sep;37(9):798-806. doi: 10.1016/j.tig.2021.03.008. Epub 2021 Apr 20. Trends Genet. 2021. PMID: 33892959 Review.
-
Genomic footprinting.Nat Methods. 2016 Mar;13(3):213-21. doi: 10.1038/nmeth.3768. Nat Methods. 2016. PMID: 26914205 Review.
Cited by
-
Beyond assembly: the increasing flexibility of single-molecule sequencing technology.Nat Rev Genet. 2023 Sep;24(9):627-641. doi: 10.1038/s41576-023-00600-1. Epub 2023 May 9. Nat Rev Genet. 2023. PMID: 37161088 Free PMC article. Review.
-
Decreased complement 4d increases poor prognosis in patients with non-small cell lung cancer combined with gastrointestinal lymph node metastasis.Exp Ther Med. 2022 Jul 5;24(3):560. doi: 10.3892/etm.2022.11497. eCollection 2022 Sep. Exp Ther Med. 2022. PMID: 35978919 Free PMC article.
-
Up-regulation of PRKDC was associated with poor renal dysfunction after renal transplantation: A multi-centre analysis.J Cell Mol Med. 2023 May;27(10):1362-1372. doi: 10.1111/jcmm.17737. Epub 2023 Apr 1. J Cell Mol Med. 2023. PMID: 37002788 Free PMC article.
-
Identification of transcription factor co-binding patterns with non-negative matrix factorization.Nucleic Acids Res. 2024 Oct 14;52(18):e85. doi: 10.1093/nar/gkae743. Nucleic Acids Res. 2024. PMID: 39217462 Free PMC article.
-
cBAF generates subnucleosomes that expand OCT4 binding and function beyond DNA motifs at enhancers.Nat Struct Mol Biol. 2024 Nov;31(11):1756-1768. doi: 10.1038/s41594-024-01344-0. Epub 2024 Jul 2. Nat Struct Mol Biol. 2024. PMID: 38956169
References
-
- Raha D, Hong M, Snyder M. ChIP-Seq: A Method for Global Identification of Regulatory Elements in the Genome. Current Protocols in Molecular Biology. 2010;91 - PubMed
-
- Reiter F, Wienerroither S, Stark A. Combinatorial function of transcription factors and cofactors. Current Opinion in Genetics & Development. 2017;43:73–81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
