

Suppression of inflammatory arthritis by the parasitic worm product ES-62 is associated with epigenetic changes in synovial fibroblasts
- PMID: 34748611
- PMCID: PMC8601611
- DOI: 10.1371/journal.ppat.1010069
Suppression of inflammatory arthritis by the parasitic worm product ES-62 is associated with epigenetic changes in synovial fibroblasts
Abstract
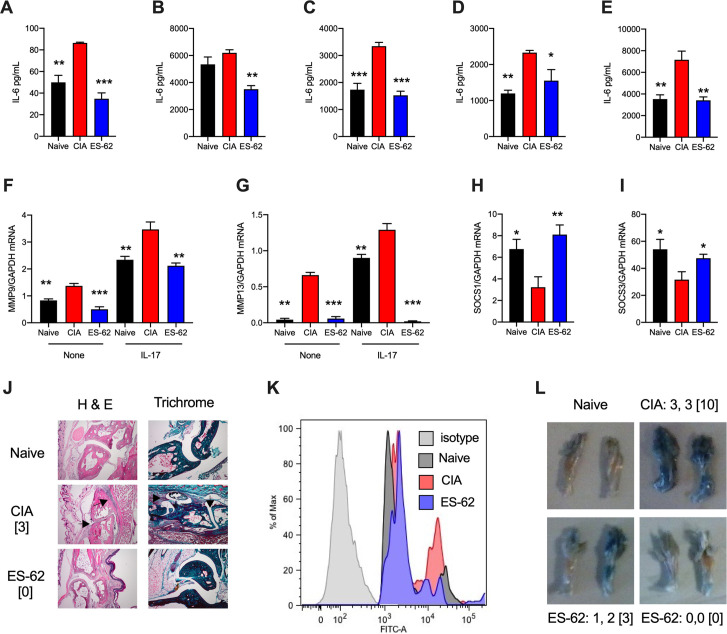
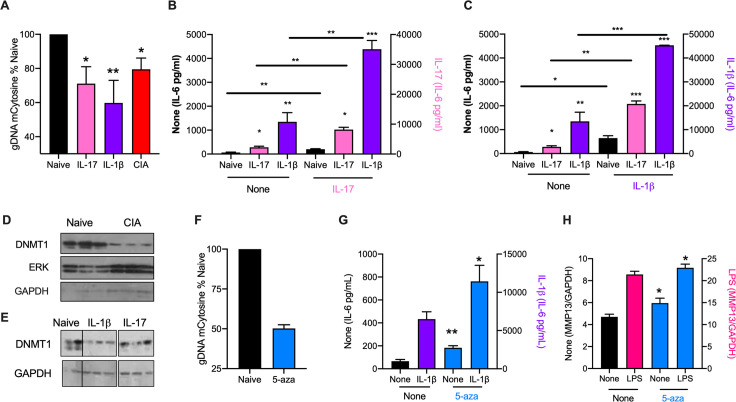
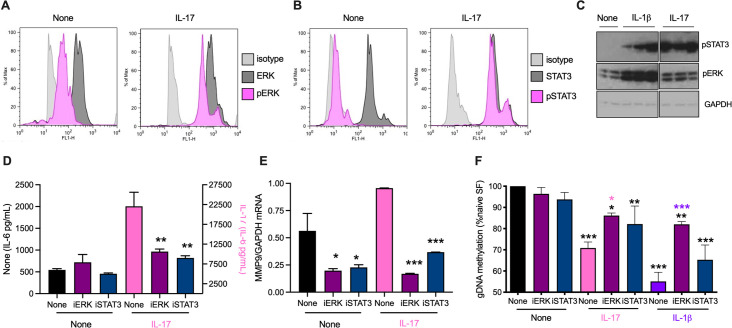
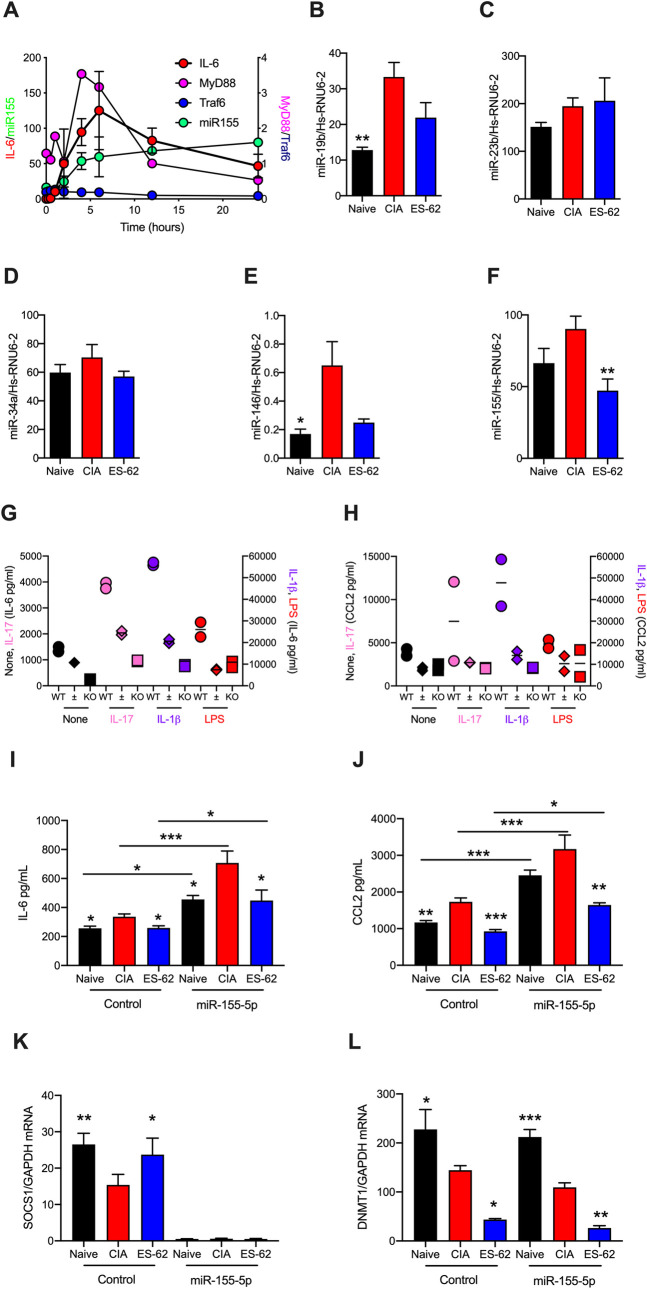
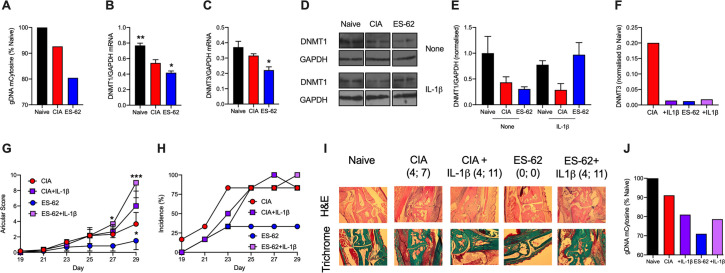
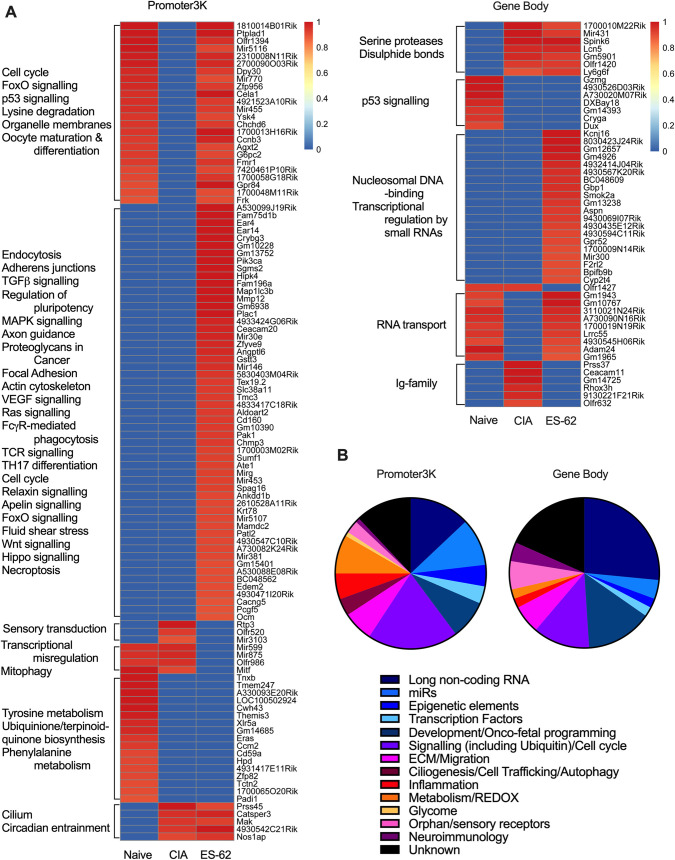
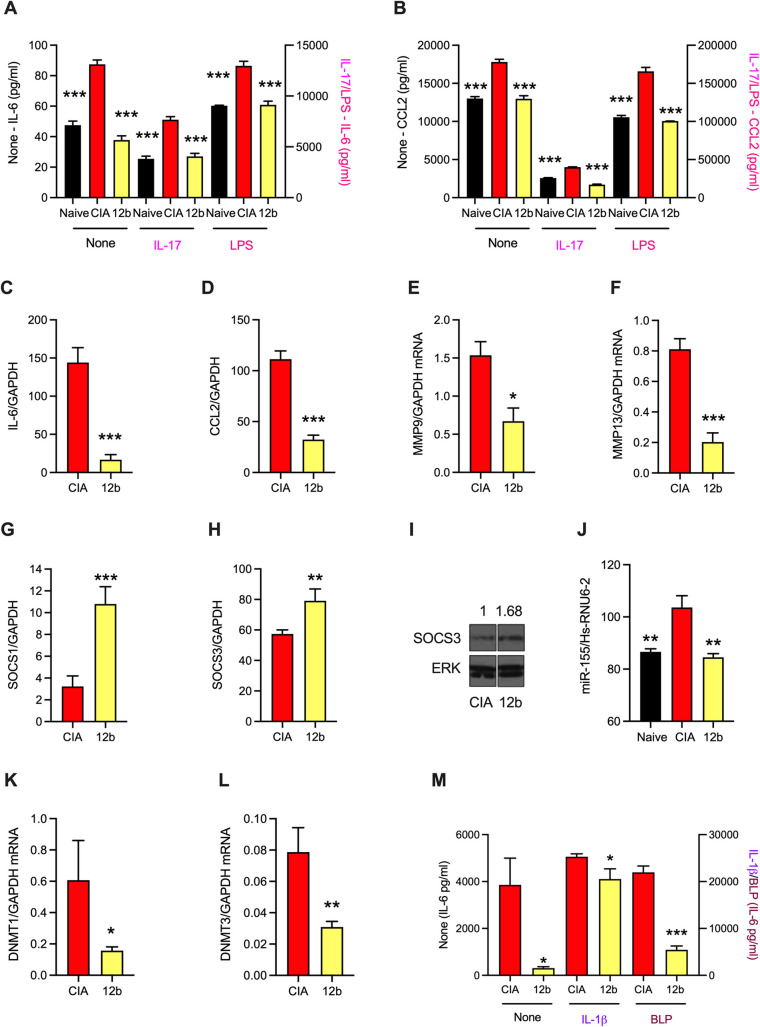
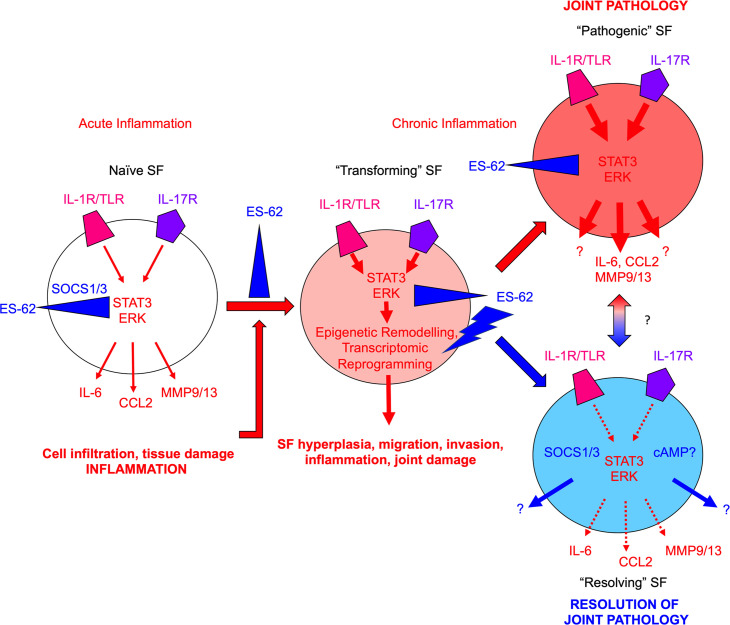
ES-62 is the major secreted protein of the parasitic filarial nematode, Acanthocheilonema viteae. The molecule exists as a large tetramer (MW, ~240kD), which possesses immunomodulatory properties by virtue of multiple phosphorylcholine (PC) moieties attached to N-type glycans. By suppressing inflammatory immune responses, ES-62 can prevent disease development in certain mouse models of allergic and autoimmune conditions, including joint pathology in collagen-induced arthritis (CIA), a model of rheumatoid arthritis (RA). Such protection is associated with functional suppression of "pathogenic" hyper-responsive synovial fibroblasts (SFs), which exhibit an aggressive inflammatory and bone-damaging phenotype induced by their epigenetic rewiring in response to the inflammatory microenvironment of the arthritic joint. Critically, exposure to ES-62 in vivo induces a stably-imprinted CIA-SF phenotype that exhibits functional responses more typical of healthy, Naïve-SFs. Consistent with this, ES-62 "rewiring" of SFs away from the hyper-responsive phenotype is associated with suppression of ERK activation, STAT3 activation and miR-155 upregulation, signals widely associated with SF pathogenesis. Surprisingly however, DNA methylome analysis of Naïve-, CIA- and ES-62-CIA-SF cohorts reveals that rather than simply preventing pathogenic rewiring of SFs, ES-62 induces further changes in DNA methylation under the inflammatory conditions pertaining in the inflamed joint, including targeting genes associated with ciliogenesis, to programme a novel "resolving" CIA-SF phenotype. In addition to introducing a previously unsuspected aspect of ES-62's mechanism of action, such unique behaviour signposts the potential for developing DNA methylation signatures predictive of pathogenesis and its resolution and hence, candidate mechanisms by which novel therapeutic interventions could prevent SFs from perpetuating joint inflammation and destruction in RA. Pertinent to these translational aspects of ES-62-behavior, small molecule analogues (SMAs) based on ES-62's active PC-moieties mimic the rewiring of SFs as well as the protection against joint disease in CIA afforded by the parasitic worm product.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Protection Against Arthritis by the Parasitic Worm Product ES-62, and Its Drug-Like Small Molecule Analogues, Is Associated With Inhibition of Osteoclastogenesis.Front Immunol. 2018 May 14;9:1016. doi: 10.3389/fimmu.2018.01016. eCollection 2018. Front Immunol. 2018. PMID: 29867986 Free PMC article.
-
Protection against collagen-induced arthritis in mice afforded by the parasitic worm product, ES-62, is associated with restoration of the levels of interleukin-10-producing B cells and reduced plasma cell infiltration of the joints.Immunology. 2014 Mar;141(3):457-66. doi: 10.1111/imm.12208. Immunology. 2014. PMID: 24708419 Free PMC article.
-
Failure of the Anti-Inflammatory Parasitic Worm Product ES-62 to Provide Protection in Mouse Models of Type I Diabetes, Multiple Sclerosis, and Inflammatory Bowel Disease.Molecules. 2018 Oct 17;23(10):2669. doi: 10.3390/molecules23102669. Molecules. 2018. PMID: 30336585 Free PMC article.
-
From the worm to the pill, the parasitic worm product ES-62 raises new horizons in the treatment of rheumatoid arthritis.Lupus. 2015 Apr;24(4-5):400-11. doi: 10.1177/0961203314560004. Lupus. 2015. PMID: 25801883 Review.
-
The anti-inflammatory potential of the filarial nematode secreted product, ES-62.Curr Top Med Chem. 2004;4(5):553-9. doi: 10.2174/1568026043451212. Curr Top Med Chem. 2004. PMID: 14965306 Review.
Cited by
-
Trichinella spiralis Paramyosin Alleviates Collagen-Induced Arthritis in Mice by Modulating CD4+ T Cell Differentiation.Int J Mol Sci. 2024 Jun 18;25(12):6706. doi: 10.3390/ijms25126706. Int J Mol Sci. 2024. PMID: 38928413 Free PMC article.
-
MicroRNAs (miRNAs) in Cardiovascular Complications of Rheumatoid Arthritis (RA): What Is New?Int J Mol Sci. 2022 May 8;23(9):5254. doi: 10.3390/ijms23095254. Int J Mol Sci. 2022. PMID: 35563643 Free PMC article. Review.
-
The parasitic worm product ES-62 protects the osteoimmunology axis in a mouse model of obesity-accelerated ageing.Front Immunol. 2022 Aug 29;13:953053. doi: 10.3389/fimmu.2022.953053. eCollection 2022. Front Immunol. 2022. PMID: 36105811 Free PMC article.
-
Immunosuppressive Ability of Trichinella spiralis Adults Can Ameliorate Type 2 Inflammation in a Murine Allergy Model.J Infect Dis. 2024 Apr 12;229(4):1215-1228. doi: 10.1093/infdis/jiad518. J Infect Dis. 2024. PMID: 38016013 Free PMC article.
-
Synovial Fibroblast Sialylation Regulates Cell Migration and Activation of Inflammatory Pathways in Arthritogenesis.Front Immunol. 2022 Mar 18;13:847581. doi: 10.3389/fimmu.2022.847581. eCollection 2022. Front Immunol. 2022. PMID: 35371069 Free PMC article.
References
-
- Steenvoorden MM, Tolboom TC, van der Pluijm G, Löwik C, Visser CP, DeGroot J, et al.. Transition of healthy to diseased synovial tissue in rheumatoid arthritis is associated with gain of mesenchymal/fibrotic characteristics. Arthritis Research & Therapy. 2006;8(6):R165. doi: 10.1186/ar2073 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
