

Pulmonary fibrosis from molecular mechanisms to therapeutic interventions: lessons from post-COVID-19 patients
- PMID: 34687672
- PMCID: PMC8546906
- DOI: 10.1016/j.bcp.2021.114812
Pulmonary fibrosis from molecular mechanisms to therapeutic interventions: lessons from post-COVID-19 patients
Abstract
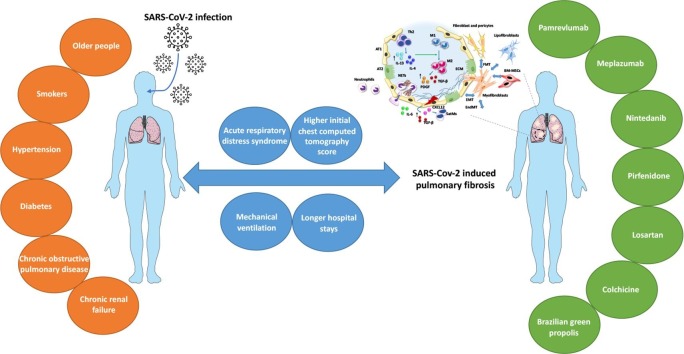
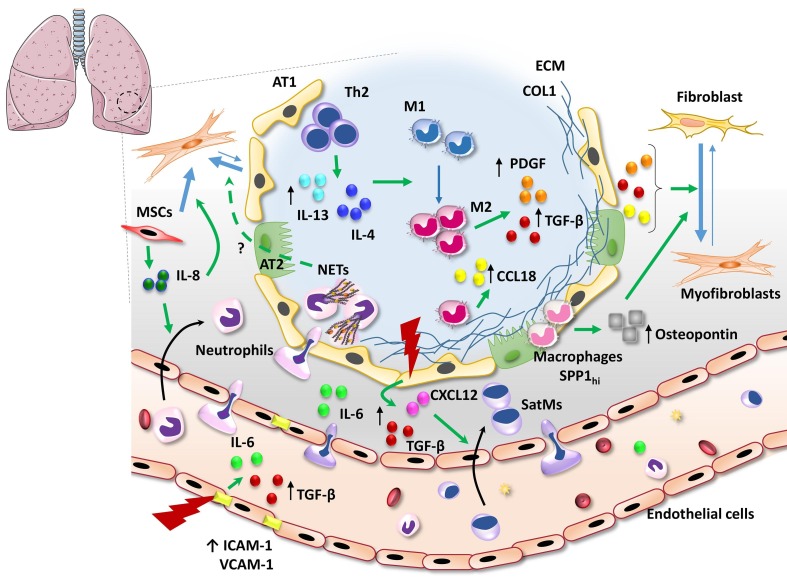
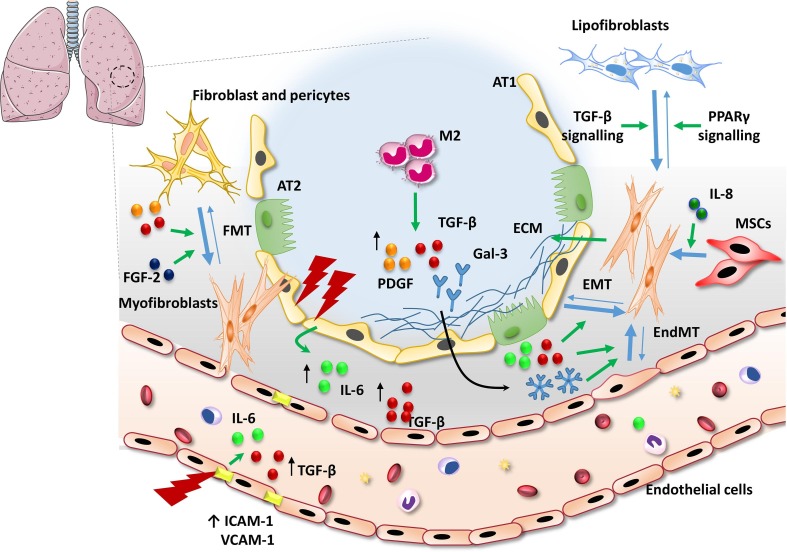
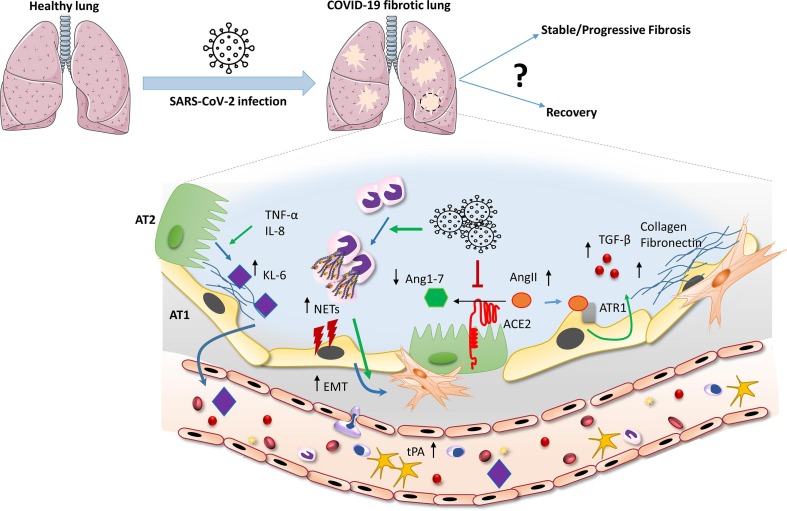
Pulmonary fibrosis (PF) is characterised by several grades of chronic inflammation and collagen deposition in the interalveolar space and is a hallmark of interstitial lung diseases (ILDs). Recently, infectious agents have emerged as driving causes for PF development; however, the role of viral/bacterial infections in the initiation and propagation of PF is still debated. In this context, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the current coronavirus disease 2019 (COVID-19) pandemic, has been associated with acute respiratory distress syndrome (ARDS) and PF development. Although the infection by SARS-CoV-2 can be eradicated in most cases, the development of fibrotic lesions cannot be precluded; furthermore, whether these lesions are stable or progressive fibrotic events is still unknown. Herein, an overview of the main molecular mechanisms driving the fibrotic process together with the currently approved and newly proposed therapeutic solutions was given. Then, the most recent data that emerged from post-COVID-19 patients was discussed, in order to compare PF and COVID-19-dependent PF, highlighting shared and specific mechanisms. A better understanding of PF aetiology is certainly needed, also to develop effective therapeutic strategies and COVID-19 pathology is offering one more chance to do it. Overall, the work reported here could help to define new approaches for therapeutic intervention in the diversity of the ILD spectrum.
Keywords: Interstitial lung disease; Myofibroblast; Pulmonary fibrosis; SARS-CoV-2 severe acute respiratory syndrome coronavirus 2.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9.Int J Mol Sci. 2022 Jul 26;23(15):8210. doi: 10.3390/ijms23158210. Int J Mol Sci. 2022. PMID: 35897786 Free PMC article. Review.
-
Pulmonary fibrosis in COVID-19: mechanisms, consequences and targets.QJM. 2023 Oct 6;116(9):750-754. doi: 10.1093/qjmed/hcad092. QJM. 2023. PMID: 37191984 Review.
-
[Pulmonary fibrosis in patients with COVID-19: A review].Ter Arkh. 2022 Dec 26;94(11):1333-1339. doi: 10.26442/00403660.2022.11.201943. Ter Arkh. 2022. PMID: 37167174 Review. Russian.
-
Overactive Epidermal Growth Factor Receptor Signaling Leads to Increased Fibrosis after Severe Acute Respiratory Syndrome Coronavirus Infection.J Virol. 2017 May 26;91(12):e00182-17. doi: 10.1128/JVI.00182-17. Print 2017 Jun 15. J Virol. 2017. PMID: 28404843 Free PMC article.
-
Involvement of the ACE2/Ang-(1-7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19.Int J Mol Sci. 2021 Nov 30;22(23):12955. doi: 10.3390/ijms222312955. Int J Mol Sci. 2021. PMID: 34884756 Free PMC article. Review.
Cited by
-
The Effect of Substrate Properties on Cellular Behavior and Nanoparticle Uptake in Human Fibroblasts and Epithelial Cells.Nanomaterials (Basel). 2024 Feb 10;14(4):342. doi: 10.3390/nano14040342. Nanomaterials (Basel). 2024. PMID: 38392715 Free PMC article.
-
Hyaluronan in the pathogenesis of acute and post-acute COVID-19 infection.Matrix Biol. 2023 Feb;116:49-66. doi: 10.1016/j.matbio.2023.02.001. Epub 2023 Feb 5. Matrix Biol. 2023. PMID: 36750167 Free PMC article. Review.
-
Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9.Int J Mol Sci. 2022 Jul 26;23(15):8210. doi: 10.3390/ijms23158210. Int J Mol Sci. 2022. PMID: 35897786 Free PMC article. Review.
-
The novel molecular mechanism of pulmonary fibrosis: insight into lipid metabolism from reanalysis of single-cell RNA-seq databases.Lipids Health Dis. 2024 Apr 3;23(1):98. doi: 10.1186/s12944-024-02062-8. Lipids Health Dis. 2024. PMID: 38570797 Free PMC article. Review.
-
Antifibrotic Drugs against Idiopathic Pulmonary Fibrosis and Pulmonary Fibrosis Induced by COVID-19: Therapeutic Approaches and Potential Diagnostic Biomarkers.Int J Mol Sci. 2024 Jan 26;25(3):1562. doi: 10.3390/ijms25031562. Int J Mol Sci. 2024. PMID: 38338840 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
