

New Challenges Resulting From the Loss of Function of Nav1.4 in Neuromuscular Diseases
- PMID: 34671263
- PMCID: PMC8521073
- DOI: 10.3389/fphar.2021.751095
New Challenges Resulting From the Loss of Function of Nav1.4 in Neuromuscular Diseases
Abstract
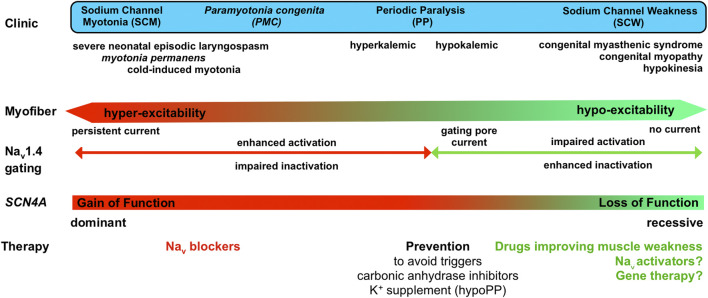
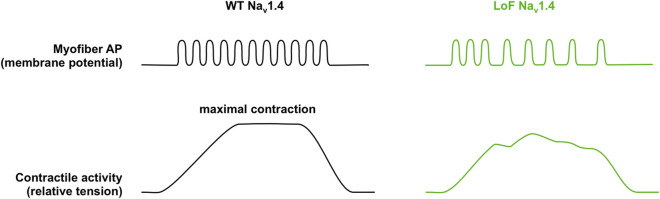
The voltage-gated sodium channel Nav1.4 is a major actor in the excitability of skeletal myofibers, driving the muscle force in response to nerve stimulation. Supporting further this key role, mutations in SCN4A, the gene encoding the pore-forming α subunit of Nav1.4, are responsible for a clinical spectrum of human diseases ranging from muscle stiffness (sodium channel myotonia, SCM) to muscle weakness. For years, only dominantly-inherited diseases resulting from Nav1.4 gain of function (GoF) were known, i.e., non-dystrophic myotonia (delayed muscle relaxation due to myofiber hyperexcitability), paramyotonia congenita and hyperkalemic or hypokalemic periodic paralyses (episodic flaccid muscle weakness due to transient myofiber hypoexcitability). These last 5 years, SCN4A mutations inducing Nav1.4 loss of function (LoF) were identified as the cause of dominantly and recessively-inherited disorders with muscle weakness: periodic paralyses with hypokalemic attacks, congenital myasthenic syndromes and congenital myopathies. We propose to name this clinical spectrum sodium channel weakness (SCW) as the mirror of SCM. Nav1.4 LoF as a cause of permanent muscle weakness was quite unexpected as the Na+ current density in the sarcolemma is large, securing the ability to generate and propagate muscle action potentials. The properties of SCN4A LoF mutations are well documented at the channel level in cellular electrophysiological studies However, much less is known about the functional consequences of Nav1.4 LoF in skeletal myofibers with no available pertinent cell or animal models. Regarding the therapeutic issues for Nav1.4 channelopathies, former efforts were aimed at developing subtype-selective Nav channel antagonists to block myofiber hyperexcitability. Non-selective, Nav channel blockers are clinically efficient in SCM and paramyotonia congenita, whereas patient education and carbonic anhydrase inhibitors are helpful to prevent attacks in periodic paralyses. Developing therapeutic tools able to counteract Nav1.4 LoF in skeletal muscles is then a new challenge in the field of Nav channelopathies. Here, we review the current knowledge regarding Nav1.4 LoF and discuss the possible therapeutic strategies to be developed in order to improve muscle force in SCW.
Keywords: congenital myasthenic syndrome (CMS); congenital myopathy (CM); loss of function; skeletal muscle; sodium channel; therapeutics.
Copyright © 2021 Nicole and Lory.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Changes of Resurgent Na+ Currents in the Nav1.4 Channel Resulting from an SCN4A Mutation Contributing to Sodium Channel Myotonia.Int J Mol Sci. 2020 Apr 8;21(7):2593. doi: 10.3390/ijms21072593. Int J Mol Sci. 2020. PMID: 32276507 Free PMC article.
-
Clinical and Molecular Spectrum of Myotonia and Periodic Paralyses Associated With Mutations in SCN4A in a Large Cohort of Italian Patients.Front Neurol. 2020 Jul 29;11:646. doi: 10.3389/fneur.2020.00646. eCollection 2020. Front Neurol. 2020. PMID: 32849172 Free PMC article.
-
Sodium Channelopathies of Skeletal Muscle.Handb Exp Pharmacol. 2018;246:309-330. doi: 10.1007/164_2017_52. Handb Exp Pharmacol. 2018. PMID: 28939973 Free PMC article. Review.
-
N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita.J Physiol. 2017 Nov 15;595(22):6837-6850. doi: 10.1113/JP274877. Epub 2017 Oct 15. J Physiol. 2017. PMID: 28940424 Free PMC article.
-
Pediatric neuromuscular channelopathies.Handb Clin Neurol. 2024;203:111-122. doi: 10.1016/B978-0-323-90820-7.00011-2. Handb Clin Neurol. 2024. PMID: 39174243 Review.
Cited by
-
Novel compound heterozygous mutations in SCN4A as a potential genetic cause contributing to myopathic manifestations: A case report and literature review.Heliyon. 2024 Mar 22;10(7):e28684. doi: 10.1016/j.heliyon.2024.e28684. eCollection 2024 Apr 15. Heliyon. 2024. PMID: 38571618 Free PMC article.
-
Activation of Voltage-Gated Na+ Current by GV-58, a Known Activator of CaV Channels.Biomedicines. 2022 Mar 20;10(3):721. doi: 10.3390/biomedicines10030721. Biomedicines. 2022. PMID: 35327523 Free PMC article.
-
The Action Potential Clamp Technique as a Tool for Risk Stratification of Sinus Bradycardia Due to Loss-of-Function Mutations in HCN4: An In Silico Exploration Based on In Vitro and In Vivo Data.Biomedicines. 2023 Sep 2;11(9):2447. doi: 10.3390/biomedicines11092447. Biomedicines. 2023. PMID: 37760888 Free PMC article.
-
Mendelian inheritance revisited: dominance and recessiveness in medical genetics.Nat Rev Genet. 2023 Jul;24(7):442-463. doi: 10.1038/s41576-023-00574-0. Epub 2023 Feb 20. Nat Rev Genet. 2023. PMID: 36806206 Review.
-
Current state of the epilepsy drug and device pipeline.Epilepsia. 2024 Apr;65(4):833-845. doi: 10.1111/epi.17884. Epub 2024 Feb 12. Epilepsia. 2024. PMID: 38345387 Review.
References
-
- Awad S. S., Lightowlers R. N., Young C., Chrzanowska-Lightowlers Z. M., Lomo T., Slater C. R. (2001). Sodium Channel mRNAs at the Neuromuscular Junction: Distinct Patterns of Accumulation and Effects of Muscle Activity. J. Neurosci. 21, 8456–8463. 10.1523/jneurosci.21-21-08456.2001 - DOI - PMC - PubMed
-
- Bailey S. J., Stocksley M. A., Buckel A., Young C., Slater C. R. (2003). Voltage-gated Sodium Channels and AnkyrinG Occupy a Different Postsynaptic Domain from Acetylcholine Receptors from an Early Stage of Neuromuscular Junction Maturation in Rats. J. Neurosci. 23, 2102–2111. 10.1523/jneurosci.23-06-02102.2003 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources