

Opposite Effects of Apoptotic and Necroptotic Cellular Pathways on Rotavirus Replication
- PMID: 34668777
- PMCID: PMC8754216
- DOI: 10.1128/JVI.01222-21
Opposite Effects of Apoptotic and Necroptotic Cellular Pathways on Rotavirus Replication
Abstract
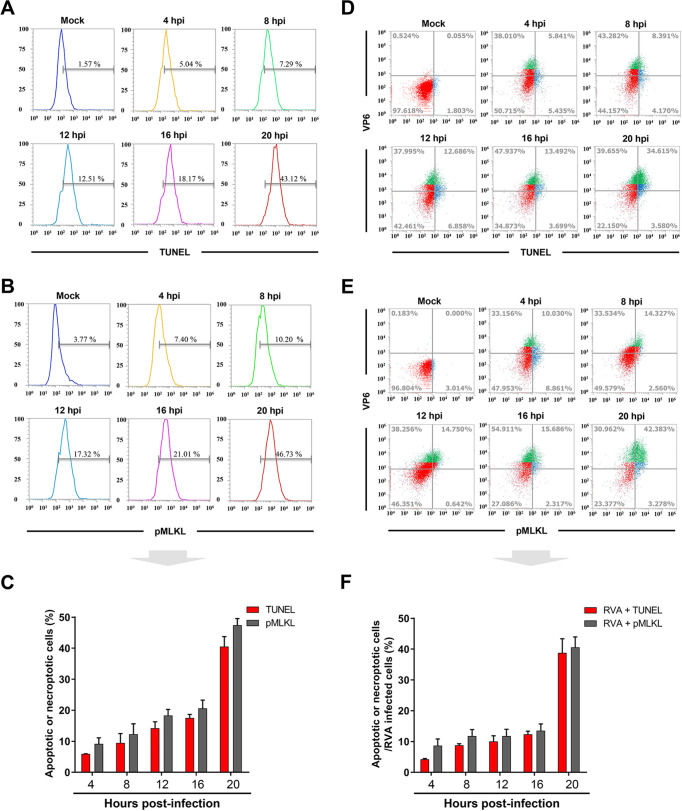
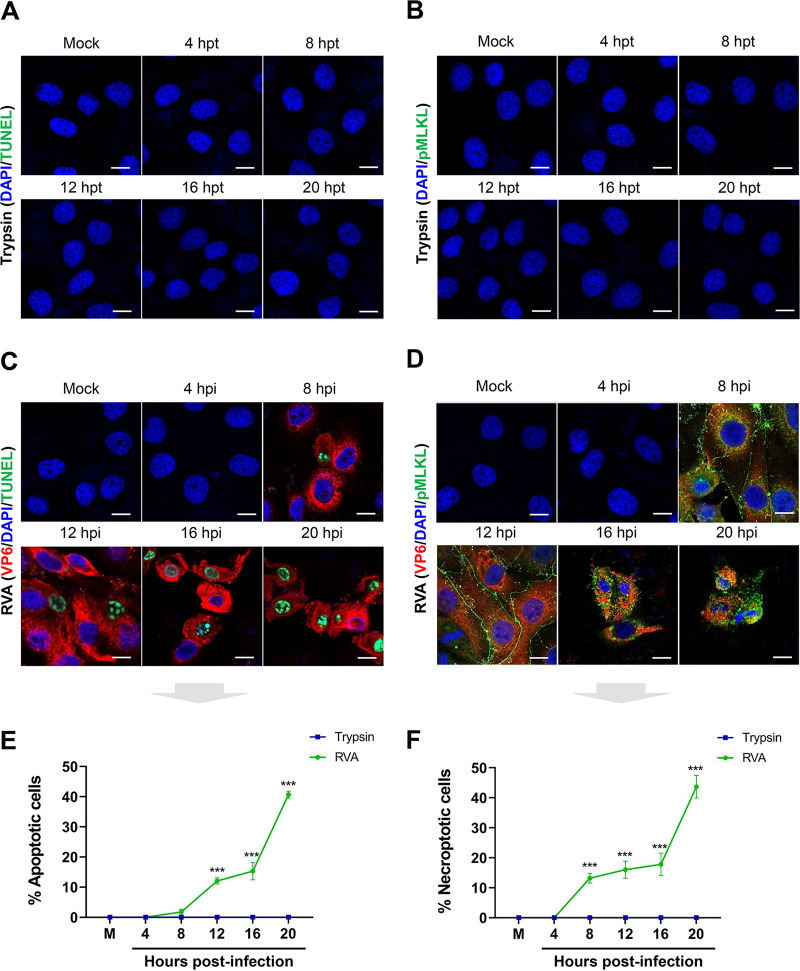
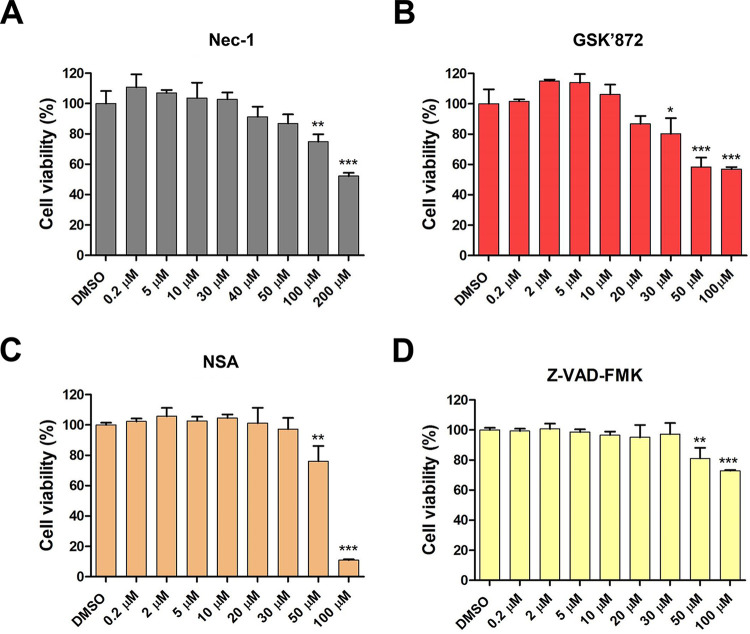
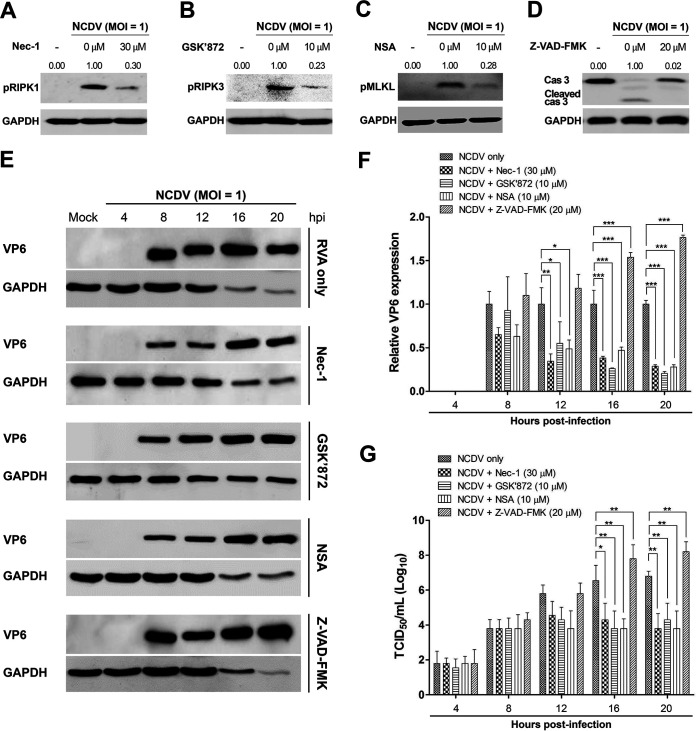
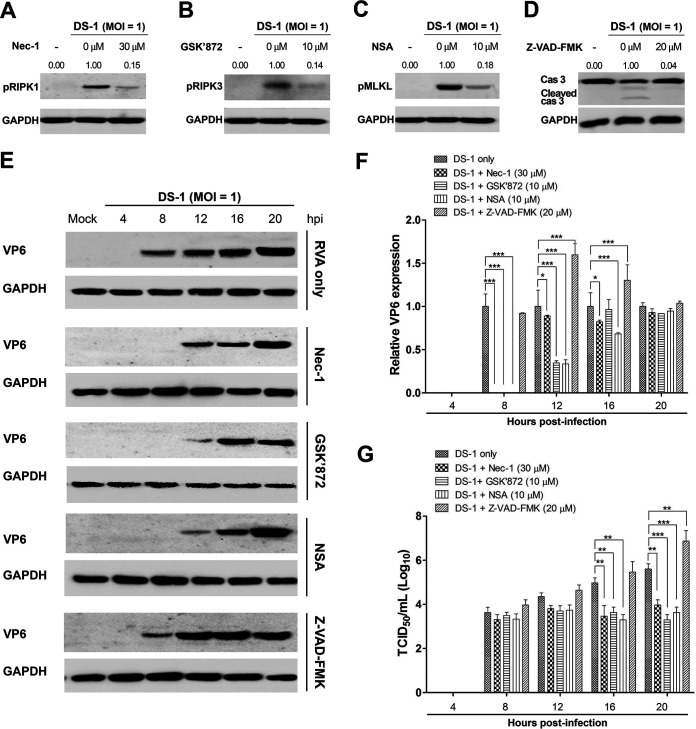
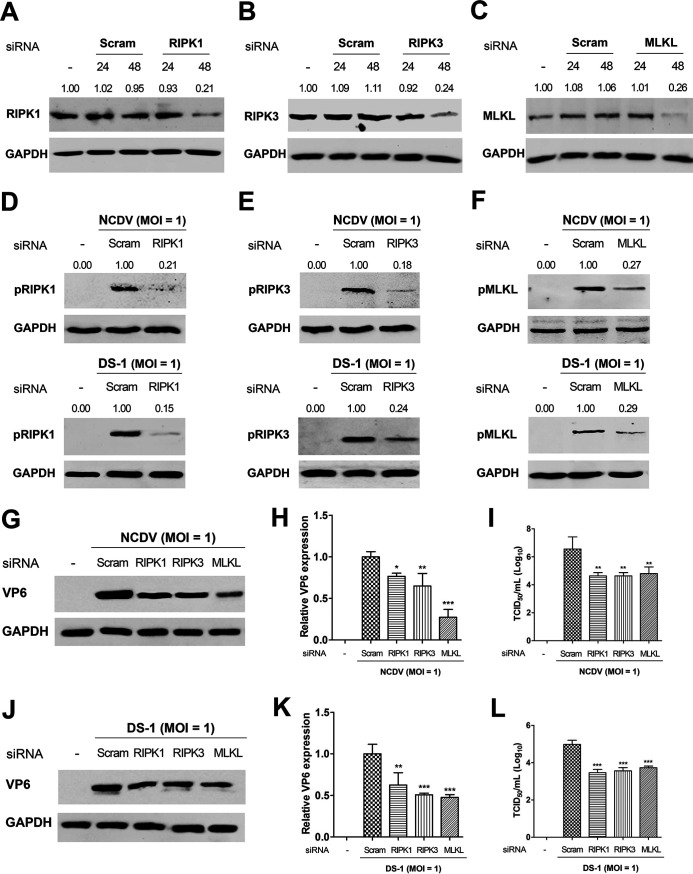
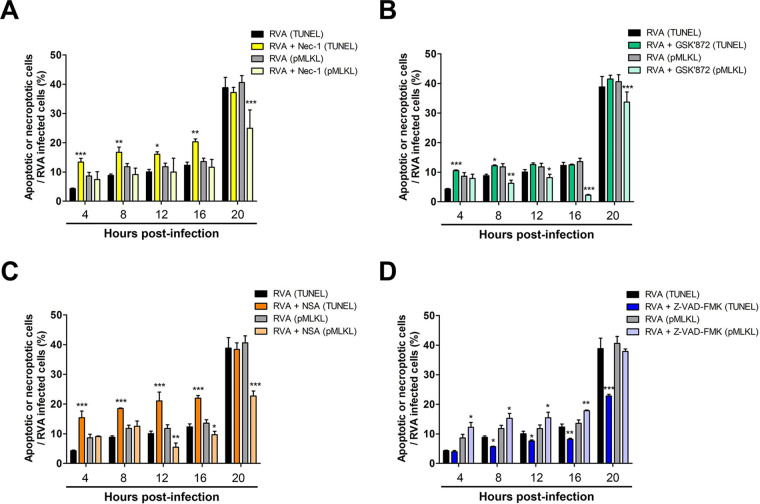
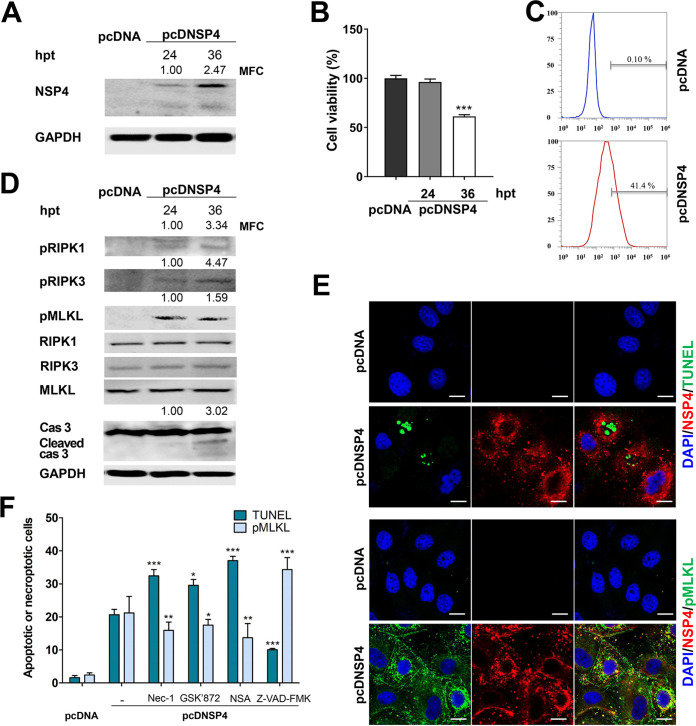
Group A rotavirus (RVA), one of the leading pathogens causing severe acute gastroenteritis in children and a wide variety of young animals worldwide, induces apoptosis upon infecting cells. Though RVA-induced apoptosis mediated via the dual modulation of its NSP4 and NSP1 proteins is relatively well studied, the nature and signaling pathway(s) involved in RVA-induced necroptosis are yet to be fully elucidated. Here, we demonstrate the nature of RVA-induced necroptosis, the signaling cascade involved, and correlation with RVA-induced apoptosis. Infection with the bovine NCDV and human DS-1 RVA strains was shown to activate receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed-lineage kinase domain-like protein (MLKL), the key necroptosis molecules in virus-infected cells. Using an immunoprecipitation assay, RIPK1 was found to bind phosphorylated RIPK3 (pRIPK3) and pMLKL. pMLKL, the major executioner molecule in the necroptotic pathway, was translocated to the plasma membrane of RVA-infected cells to puncture the cell membrane. Interestingly, transfection of RVA NSP4 also induced necroptosis through the RIPK1/RIPK3/MLKL necroptosis pathway. Blockage of each key necroptosis molecule in the RVA-infected or NSP4-transfected cells resulted in decreased necroptosis but increased cell viability and apoptosis, thereby resulting in decreased viral yields in the RVA-infected cells. In contrast, suppression of RVA-induced apoptosis increased necroptosis and virus yields. Our findings suggest that RVA NSP4 also induces necroptosis via the RIPK1/RIPK3/MLKL necroptosis pathway. Moreover, necroptosis and apoptosis-which have proviral and antiviral effects, respectively-exhibited cross talk in RVA-infected cells. These findings significantly increase our understanding of the nature of RVA-induced necroptosis and the cross talk between RVA-induced necroptosis and apoptosis. IMPORTANCE Viral infection usually culminates in cell death through apoptosis, necroptosis, and, rarely, pyroptosis. Necroptosis is a form of programmed necrosis that is mediated by signaling complexes of the receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed-lineage kinase domain-like protein (MLKL). Although apoptosis induction by rotavirus and its NSP4 protein is well known, rotavirus-induced necroptosis is not fully understood. Here, we demonstrate that rotavirus and also its NSP4 protein can induce necroptosis in cultured cells through activation of the RIPK1/RIPK3/MLKL necroptosis pathway. Moreover, rotavirus-induced necroptosis and apoptosis have opposite effects on viral yield, i.e., they function as proviral and antiviral processes, respectively, and counterbalance each other in rotavirus-infected cells. Our findings provide important insights for understanding the nature of rotavirus-induced necroptosis and the development of novel therapeutic strategies against infection with rotavirus and other RNA viruses.
Keywords: NSP4; RIPK1/RIPK3/MLKL; apoptosis; necroptosis; rotavirus.
Figures






Similar articles
-
Porcine sapovirus-induced RIPK1-dependent necroptosis is proviral in LLC-PK cells.PLoS One. 2023 Feb 3;18(2):e0279843. doi: 10.1371/journal.pone.0279843. eCollection 2023. PLoS One. 2023. PMID: 36735696 Free PMC article.
-
Rotavirus non-structural protein 4 usurps host cellular RIPK1-RIPK3 complex to induce MLKL-dependent necroptotic cell death.Biochim Biophys Acta Mol Cell Res. 2024 Jun;1871(5):119745. doi: 10.1016/j.bbamcr.2024.119745. Epub 2024 May 6. Biochim Biophys Acta Mol Cell Res. 2024. PMID: 38719029
-
RIPK1-RIPK3-MLKL-Associated Necroptosis Drives Leishmania infantum Killing in Neutrophils.Front Immunol. 2018 Aug 14;9:1818. doi: 10.3389/fimmu.2018.01818. eCollection 2018. Front Immunol. 2018. PMID: 30154785 Free PMC article.
-
The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis.Int Rev Cell Mol Biol. 2017;328:253-275. doi: 10.1016/bs.ircmb.2016.08.007. Epub 2016 Sep 22. Int Rev Cell Mol Biol. 2017. PMID: 28069136 Free PMC article. Review.
-
Viral-induced neuronal necroptosis: Detrimental to brain function and regulation by necroptosis inhibitors.Biochem Pharmacol. 2023 Jul;213:115591. doi: 10.1016/j.bcp.2023.115591. Epub 2023 May 16. Biochem Pharmacol. 2023. PMID: 37196683 Review.
Cited by
-
Identification and characterization of a marine bacterium extract from Mameliella sp. M20D2D8 with antiviral effects against influenza A and B viruses.Arch Virol. 2024 Feb 7;169(3):41. doi: 10.1007/s00705-024-05979-8. Arch Virol. 2024. PMID: 38326489 Free PMC article.
-
Lipidomics reveals the significance and mechanism of the cellular ceramide metabolism for rotavirus replication.J Virol. 2024 Apr 16;98(4):e0006424. doi: 10.1128/jvi.00064-24. Epub 2024 Mar 15. J Virol. 2024. PMID: 38488360 Free PMC article.
-
Porcine sapovirus-induced RIPK1-dependent necroptosis is proviral in LLC-PK cells.PLoS One. 2023 Feb 3;18(2):e0279843. doi: 10.1371/journal.pone.0279843. eCollection 2023. PLoS One. 2023. PMID: 36735696 Free PMC article.
-
Approaches to Evaluating Necroptosis in Virus-Infected Cells.Subcell Biochem. 2023;106:37-75. doi: 10.1007/978-3-031-40086-5_2. Subcell Biochem. 2023. PMID: 38159223
-
OSW-1 triggers necroptosis in colorectal cancer cells through the RIPK1/RIPK3/MLKL signaling pathway facilitated by the RIPK1-p62/SQSTM1 complex.World J Gastroenterol. 2024 Apr 21;30(15):2155-2174. doi: 10.3748/wjg.v30.i15.2155. World J Gastroenterol. 2024. PMID: 38681991 Free PMC article.
References
-
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. 2012. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19:107–120. 10.1038/cdd.2011.96. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
