

Calling large indels in 1047 Arabidopsis with IndelEnsembler
- PMID: 34643730
- PMCID: PMC8565333
- DOI: 10.1093/nar/gkab904
Calling large indels in 1047 Arabidopsis with IndelEnsembler
Abstract
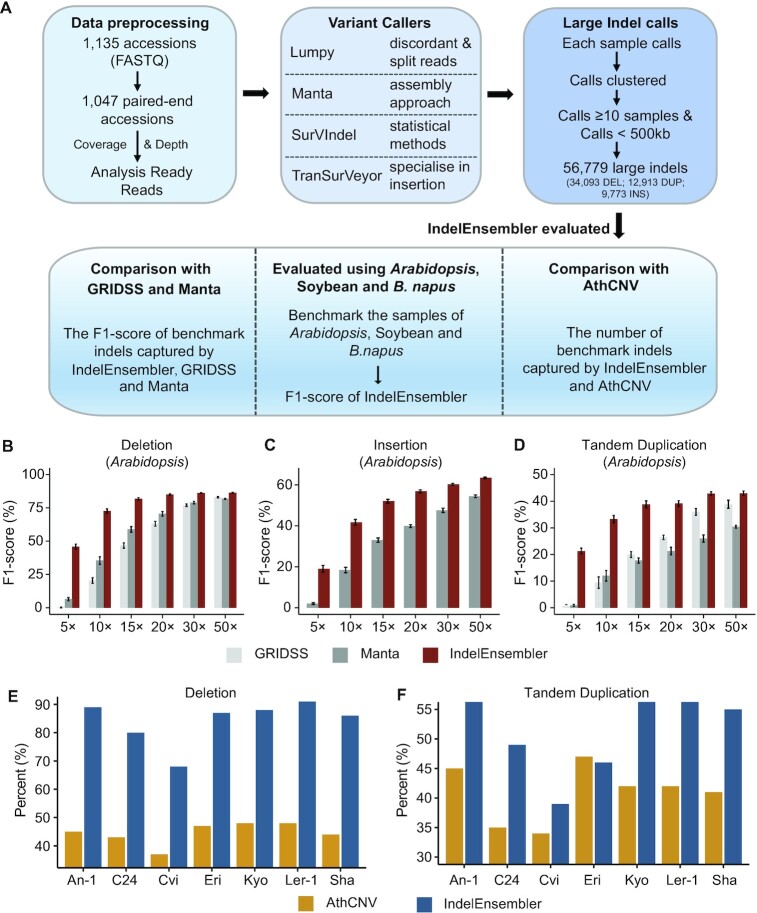
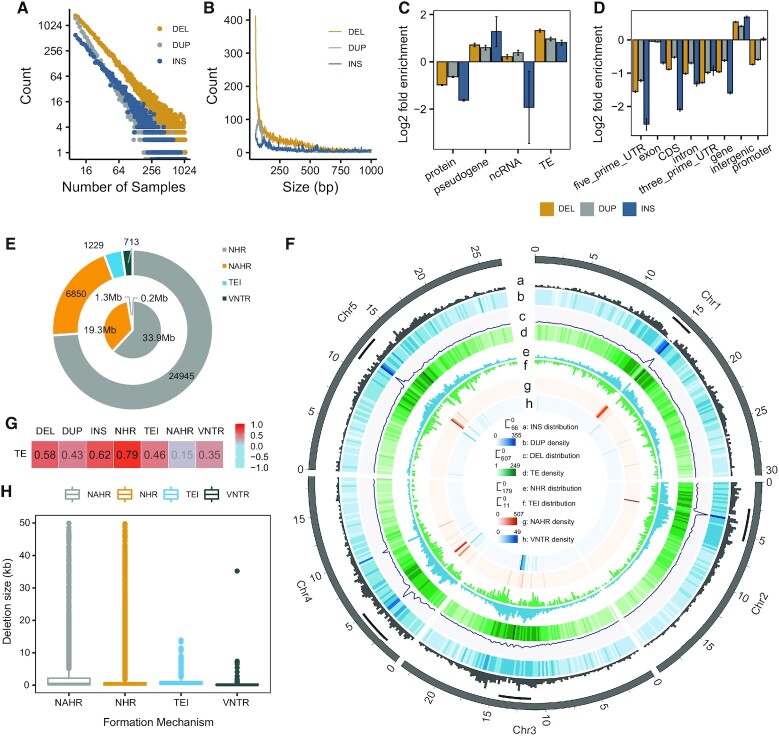
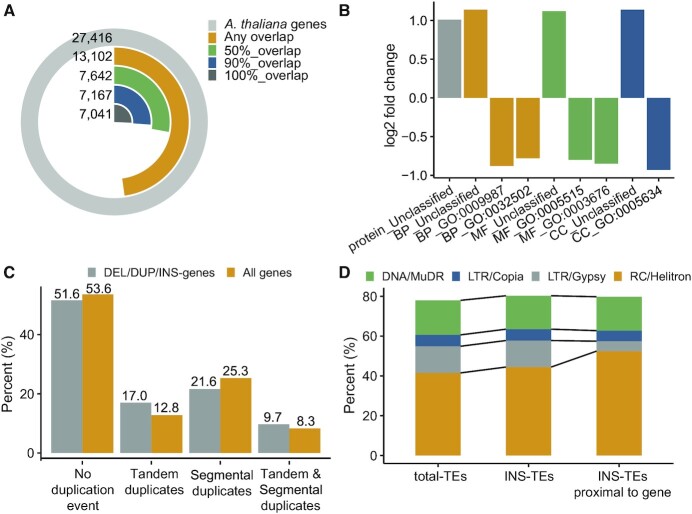
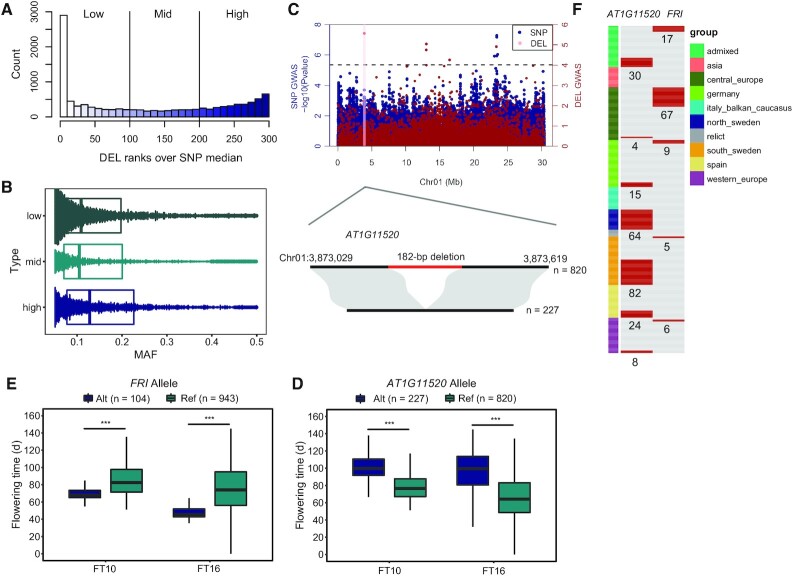
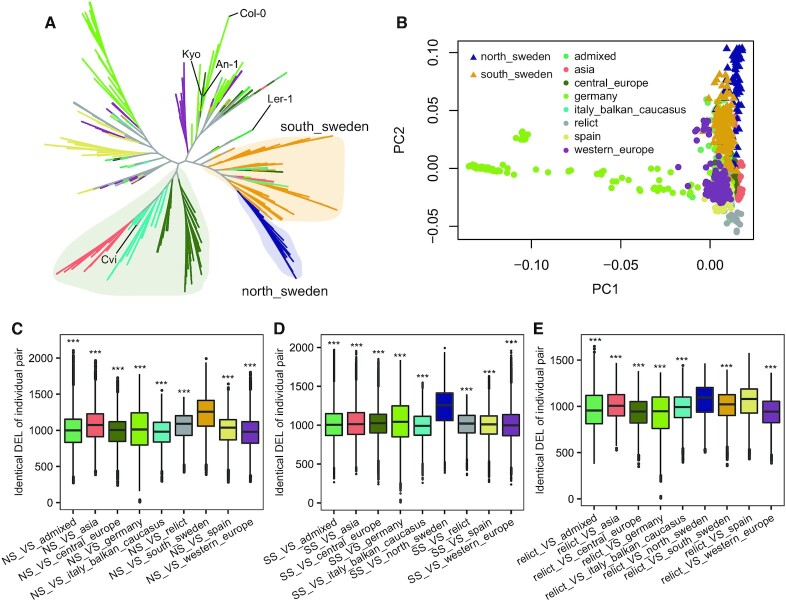
Large indels greatly impact the observable phenotypes in different organisms including plants and human. Hence, extracting large indels with high precision and sensitivity is important. Here, we developed IndelEnsembler to detect large indels in 1047 Arabidopsis whole-genome sequencing data. IndelEnsembler identified 34 093 deletions, 12 913 tandem duplications and 9773 insertions. Our large indel dataset was more comprehensive and accurate compared with the previous dataset of AthCNV (1). We captured nearly twice of the ground truth deletions and on average 27% more ground truth duplications compared with AthCNV, though our dataset has less number of large indels compared with AthCNV. Our large indels were positively correlated with transposon elements across the Arabidopsis genome. The non-homologous recombination events were the major formation mechanism of deletions in Arabidopsis genome. The Neighbor joining (NJ) tree constructed based on IndelEnsembler's deletions clearly divided the geographic subgroups of 1047 Arabidopsis. More importantly, our large indels represent a previously unassessed source of genetic variation. Approximately 49% of the deletions have low linkage disequilibrium (LD) with surrounding single nucleotide polymorphisms. Some of them could affect trait performance. For instance, using deletion-based genome-wide association study (DEL-GWAS), the accessions containing a 182-bp deletion in AT1G11520 had delayed flowering time and all accessions in north Sweden had the 182-bp deletion. We also found the accessions with 65-bp deletion in the first exon of AT4G00650 (FRI) flowered earlier than those without it. These two deletions cannot be detected in AthCNV and, interestingly, they do not co-occur in any Arabidopsis thaliana accession. By SNP-GWAS, surrounding SNPs of these two deletions do not correlate with flowering time. This example demonstrated that existing large indel datasets miss phenotypic variations and our large indel dataset filled in the gap.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

Similar articles
-
The pattern of insertion/deletion polymorphism in Arabidopsis thaliana.Mol Genet Genomics. 2008 Oct;280(4):351-61. doi: 10.1007/s00438-008-0370-1. Epub 2008 Aug 9. Mol Genet Genomics. 2008. PMID: 18690477
-
Different patterns of gene structure divergence following gene duplication in Arabidopsis.BMC Genomics. 2013 Sep 24;14:652. doi: 10.1186/1471-2164-14-652. BMC Genomics. 2013. PMID: 24063813 Free PMC article.
-
A De Novo Genome Sequence Assembly of the Arabidopsis thaliana Accession Niederzenz-1 Displays Presence/Absence Variation and Strong Synteny.PLoS One. 2016 Oct 6;11(10):e0164321. doi: 10.1371/journal.pone.0164321. eCollection 2016. PLoS One. 2016. PMID: 27711162 Free PMC article.
-
The association of insertions/deletions (INDELs) and variable number tandem repeats (VNTRs) with obesity and its related traits and complications.J Physiol Anthropol. 2017 Jun 14;36(1):25. doi: 10.1186/s40101-017-0142-x. J Physiol Anthropol. 2017. PMID: 28615046 Free PMC article. Review.
-
The repeatability of genome-wide mutation rate and spectrum estimates.Curr Genet. 2016 Aug;62(3):507-12. doi: 10.1007/s00294-016-0573-7. Epub 2016 Feb 26. Curr Genet. 2016. PMID: 26919990 Free PMC article. Review.
Cited by
-
Lint percentage and boll weight QTLs in three excellent upland cotton (Gossypium hirsutum): ZR014121, CCRI60, and EZ60.BMC Plant Biol. 2023 Apr 5;23(1):179. doi: 10.1186/s12870-023-04147-5. BMC Plant Biol. 2023. PMID: 37020180 Free PMC article.
-
Extensive sequence duplication in Arabidopsis revealed by pseudo-heterozygosity.Genome Biol. 2023 Mar 9;24(1):44. doi: 10.1186/s13059-023-02875-3. Genome Biol. 2023. PMID: 36895055 Free PMC article.
-
InDels Identification and Association Analysis with Spike and Awn Length in Chinese Wheat Mini-Core Collection.Int J Mol Sci. 2022 May 17;23(10):5587. doi: 10.3390/ijms23105587. Int J Mol Sci. 2022. PMID: 35628397 Free PMC article.
-
SVvalidation: A long-read-based validation method for genomic structural variation.PLoS One. 2024 Jan 5;19(1):e0291741. doi: 10.1371/journal.pone.0291741. eCollection 2024. PLoS One. 2024. PMID: 38181020 Free PMC article.
-
SVcnn: an accurate deep learning-based method for detecting structural variation based on long-read data.BMC Bioinformatics. 2023 May 23;24(1):213. doi: 10.1186/s12859-023-05324-x. BMC Bioinformatics. 2023. PMID: 37221476 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
