

Redox Homeostasis Involvement in the Pharmacological Effects of Metformin in Systemic Lupus Erythematosus
- PMID: 34619975
- PMCID: PMC8982129
- DOI: 10.1089/ars.2021.0070
Redox Homeostasis Involvement in the Pharmacological Effects of Metformin in Systemic Lupus Erythematosus
Abstract
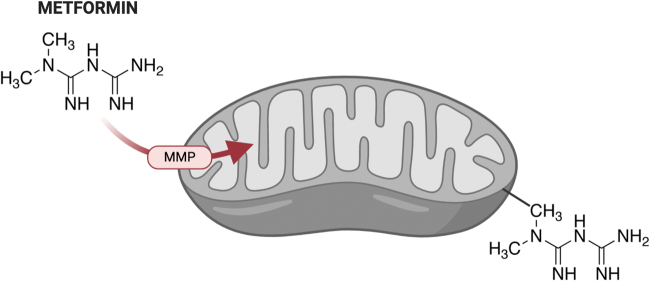
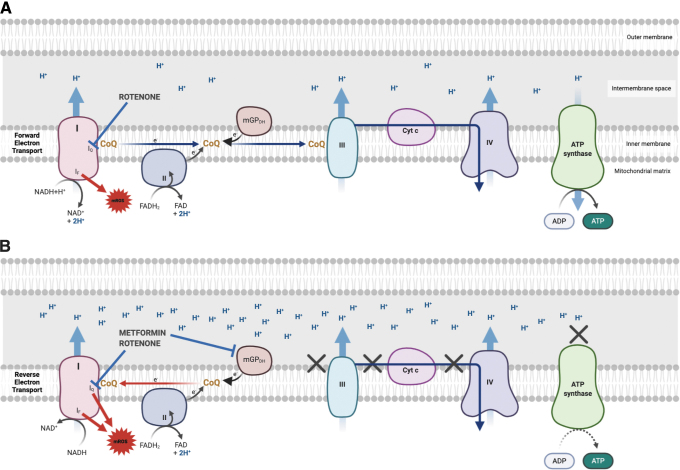
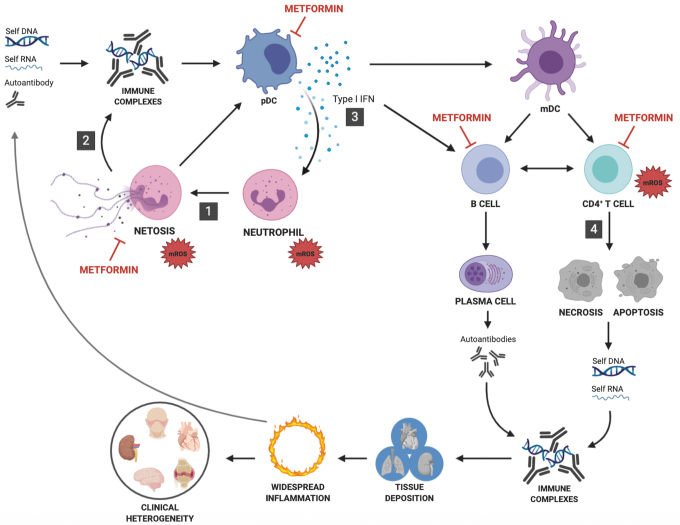
Significance: Metformin has been proposed as a treatment for systemic lupus erythematosus (SLE). The primary target of metformin, the electron transport chain complex I in the mitochondria, is associated with redox homeostasis in immune cells, which plays a critical role in the pathogenesis of autoimmune diseases. This review addresses the evidence and knowledge gaps on whether a beneficial effect of metformin in lupus may be due to a restoration of a balanced redox state. Recent Advances: Clinical trials in SLE patients with mild-to-moderate disease activity and preclinical studies in mice have provided encouraging results for metformin. The mechanism by which this therapeutic effect was achieved is largely unknown. Metformin regulates redox homeostasis in a context-specific manner. Multiple cell types contribute to SLE, with evidence of increased mitochondrial oxidative stress in T cells and neutrophils. Critical Issues: The major knowledge gaps are whether the efficacy of metformin is linked to a restored redox homeostasis in the immune system, and if it does, in which cell types it occurs? We also need to know which patients may have a better response to metformin, and whether it corresponds to a specific mechanism? Finally, the identification of biomarkers to predict treatment outcomes would be of great value. Future Directions: Mechanistic studies must address the context-dependent pharmacological effects of metformin. Multiple cell types as well as a complex disease etiology should be considered. These studies must integrate the rapid advances made in understanding how metabolic programs direct the effector functions of immune cells. Antioxid. Redox Signal. 36, 462-479.
Keywords: ROS; lupus; metformin; mitochondria.
Conflict of interest statement
No competing financial interests exist.
Figures
Similar articles
-
Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin.Arthritis Rheumatol. 2015 Dec;67(12):3190-200. doi: 10.1002/art.39296. Arthritis Rheumatol. 2015. PMID: 26245802 Clinical Trial.
-
Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis.Int J Mol Sci. 2024 Jun 3;25(11):6162. doi: 10.3390/ijms25116162. Int J Mol Sci. 2024. PMID: 38892349 Free PMC article. Review.
-
T Cell Homeostatic Proliferation Promotes a Redox State That Drives Metabolic and Epigenetic Upregulation of Inflammatory Pathways in Lupus.Antioxid Redox Signal. 2022 Mar;36(7-9):410-422. doi: 10.1089/ars.2021.0078. Epub 2021 Nov 9. Antioxid Redox Signal. 2022. PMID: 34328790 Free PMC article. Review.
-
Normalization of CD4+ T cell metabolism reverses lupus.Sci Transl Med. 2015 Feb 11;7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835. Sci Transl Med. 2015. PMID: 25673763 Free PMC article.
-
Mitochondrial Dysfunction in Systemic Lupus Erythematosus: Insights and Therapeutic Potential.Diseases. 2024 Sep 23;12(9):226. doi: 10.3390/diseases12090226. Diseases. 2024. PMID: 39329895 Free PMC article. Review.
Cited by
-
The Intersection of Cellular and Systemic Metabolism: Metabolic Syndrome in Systemic Lupus Erythematosus.Endocrinology. 2022 Jul 1;163(7):bqac067. doi: 10.1210/endocr/bqac067. Endocrinology. 2022. PMID: 35560001 Free PMC article. Review.
-
Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus.Front Immunol. 2022 Jul 25;13:929520. doi: 10.3389/fimmu.2022.929520. eCollection 2022. Front Immunol. 2022. PMID: 35958572 Free PMC article. Review.
-
Toxic Effects of Penetrating Cations.Membranes (Basel). 2023 Oct 22;13(10):841. doi: 10.3390/membranes13100841. Membranes (Basel). 2023. PMID: 37888013 Free PMC article. Review.
-
Anti-Inflammatory Potential of the Anti-Diabetic Drug Metformin in the Prevention of Inflammatory Complications and Infectious Diseases Including COVID-19: A Narrative Review.Int J Mol Sci. 2024 May 10;25(10):5190. doi: 10.3390/ijms25105190. Int J Mol Sci. 2024. PMID: 38791227 Free PMC article. Review.
-
Redox Pathogenesis in Rheumatic Diseases.ACR Open Rheumatol. 2024 Jun;6(6):334-346. doi: 10.1002/acr2.11668. Epub 2024 Apr 25. ACR Open Rheumatol. 2024. PMID: 38664977 Free PMC article. Review.
References
-
- An H, Wei R, Ke J, Yang J, Liu Y, Wang X, Wang G, and Hong T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J Diabetes Complications 30: 1017–1024, 2016. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
