

Oncogenic KRAS blockade therapy: renewed enthusiasm and persistent challenges
- PMID: 34607583
- PMCID: PMC8489073
- DOI: 10.1186/s12943-021-01422-7
Oncogenic KRAS blockade therapy: renewed enthusiasm and persistent challenges
Abstract
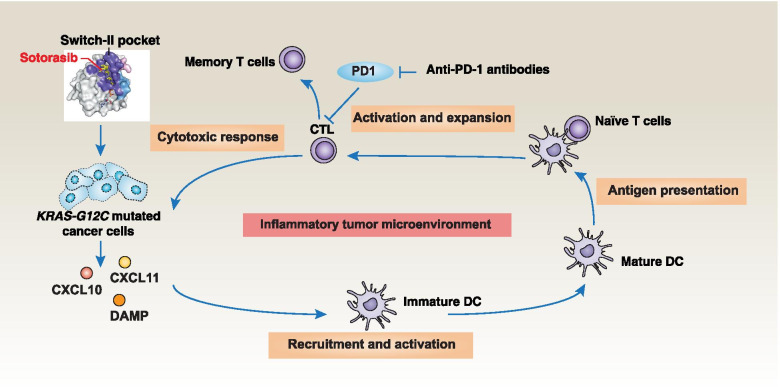
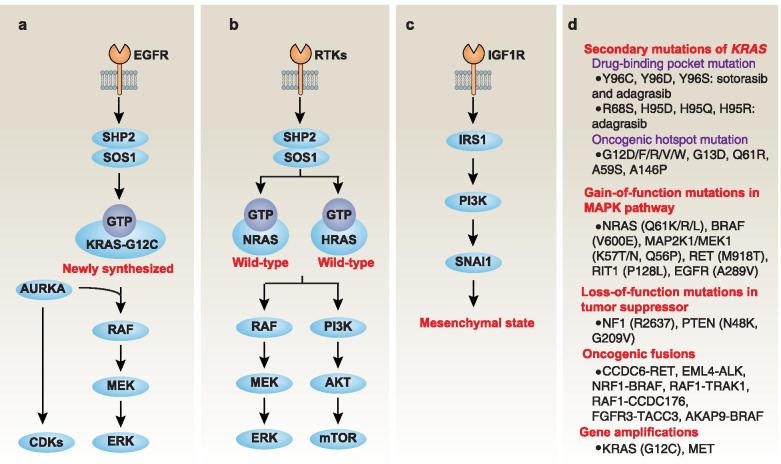
Across a broad range of human cancers, gain-of-function mutations in RAS genes (HRAS, NRAS, and KRAS) lead to constitutive activity of oncoproteins responsible for tumorigenesis and cancer progression. The targeting of RAS with drugs is challenging because RAS lacks classic and tractable drug binding sites. Over the past 30 years, this perception has led to the pursuit of indirect routes for targeting RAS expression, processing, upstream regulators, or downstream effectors. After the discovery that the KRAS-G12C variant contains a druggable pocket below the switch-II loop region, it has become possible to design irreversible covalent inhibitors for the variant with improved potency, selectivity and bioavailability. Two such inhibitors, sotorasib (AMG 510) and adagrasib (MRTX849), were recently evaluated in phase I-III trials for the treatment of non-small cell lung cancer with KRAS-G12C mutations, heralding a new era of precision oncology. In this review, we outline the mutations and functions of KRAS in human tumors and then analyze indirect and direct approaches to shut down the oncogenic KRAS network. Specifically, we discuss the mechanistic principles, clinical features, and strategies for overcoming primary or secondary resistance to KRAS-G12C blockade.
Keywords: Covalent inhibitor; Drug resistance; Gene mutation; KRAS; Targeted therapy.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests. GK is a co-founder of EverImmune, Samsara Therapeutics, and Therafast Bio.
Figures
Similar articles
-
Structural insights into small-molecule KRAS inhibitors for targeting KRAS mutant cancers.Eur J Med Chem. 2024 Nov 5;277:116771. doi: 10.1016/j.ejmech.2024.116771. Epub 2024 Aug 15. Eur J Med Chem. 2024. PMID: 39167893 Review.
-
Beyond KRAS(G12C): Biochemical and Computational Characterization of Sotorasib and Adagrasib Binding Specificity and the Critical Role of H95 and Y96.ACS Chem Biol. 2024 Oct 18;19(10):2152-2164. doi: 10.1021/acschembio.4c00315. Epub 2024 Sep 16. ACS Chem Biol. 2024. PMID: 39283696
-
KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne?Cancer Treat Rev. 2020 Mar;84:101974. doi: 10.1016/j.ctrv.2020.101974. Epub 2020 Jan 23. Cancer Treat Rev. 2020. PMID: 32014824 Free PMC article. Review.
-
Targeted Therapies for Previously "Undruggable" KRAS-Mutated Non-Small Cell Lung Cancer: A Review of Sotorasib and Adagrasib.Ann Pharmacother. 2024 Jun;58(6):622-635. doi: 10.1177/10600280231197459. Epub 2023 Sep 12. Ann Pharmacother. 2024. PMID: 37700573 Review.
-
KRAS mutation: from undruggable to druggable in cancer.Signal Transduct Target Ther. 2021 Nov 15;6(1):386. doi: 10.1038/s41392-021-00780-4. Signal Transduct Target Ther. 2021. PMID: 34776511 Free PMC article. Review.
Cited by
-
Farnesyl-transferase inhibitors show synergistic anticancer effects in combination with novel KRAS-G12C inhibitors.Br J Cancer. 2024 Apr;130(6):1059-1072. doi: 10.1038/s41416-024-02586-x. Epub 2024 Jan 26. Br J Cancer. 2024. PMID: 38278976 Free PMC article.
-
Daily Practice Assessment of KRAS Status in NSCLC Patients: A New Challenge for the Thoracic Pathologist Is Right around the Corner.Cancers (Basel). 2022 Mar 23;14(7):1628. doi: 10.3390/cancers14071628. Cancers (Basel). 2022. PMID: 35406400 Free PMC article. Review.
-
Synthetic Vulnerabilities in the KRAS Pathway.Cancers (Basel). 2022 Jun 8;14(12):2837. doi: 10.3390/cancers14122837. Cancers (Basel). 2022. PMID: 35740503 Free PMC article. Review.
-
143D, a novel selective KRASG12C inhibitor exhibits potent antitumor activity in preclinical models.Acta Pharmacol Sin. 2023 Jul;44(7):1475-1486. doi: 10.1038/s41401-023-01053-2. Epub 2023 Feb 1. Acta Pharmacol Sin. 2023. PMID: 36725884 Free PMC article.
-
Novel Inhibitor-Based Therapies for Thyroid Cancer-An Update.Int J Mol Sci. 2021 Oct 31;22(21):11829. doi: 10.3390/ijms222111829. Int J Mol Sci. 2021. PMID: 34769260 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
