

The Oncogenic Signaling Disruptor, NDRG1: Molecular and Cellular Mechanisms of Activity
- PMID: 34572031
- PMCID: PMC8465210
- DOI: 10.3390/cells10092382
The Oncogenic Signaling Disruptor, NDRG1: Molecular and Cellular Mechanisms of Activity
Abstract
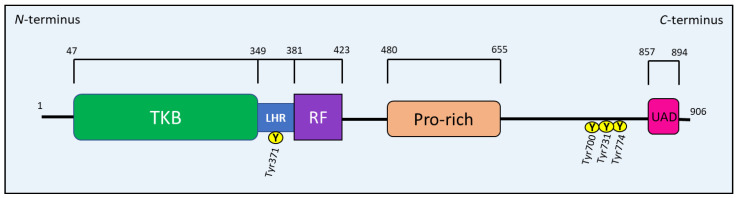
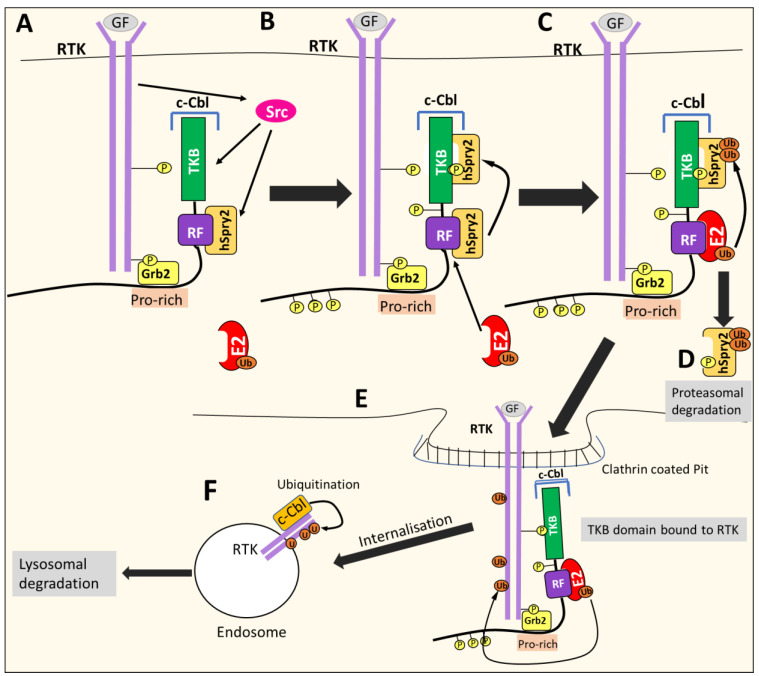
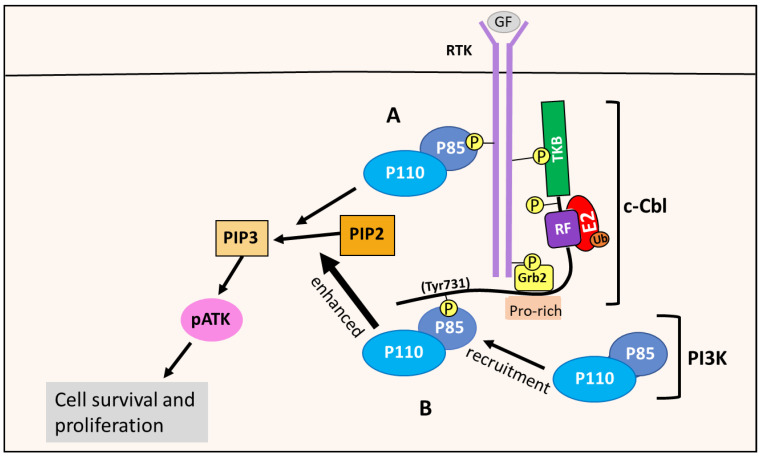
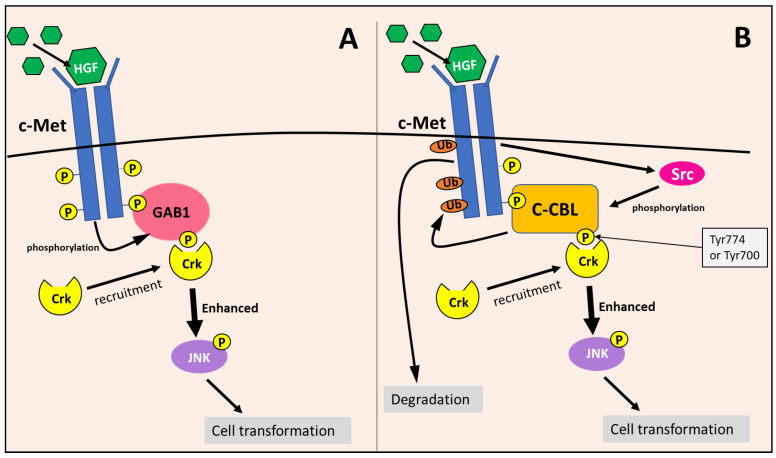
NDRG1 is an oncogenic signaling disruptor that plays a key role in multiple cancers, including aggressive pancreatic tumors. Recent studies have indicated a role for NDRG1 in the inhibition of multiple tyrosine kinases, including EGFR, c-Met, HER2 and HER3, etc. The mechanism of activity of NDRG1 remains unclear, but to impart some of its functions, NDRG1 binds directly to key effector molecules that play roles in tumor suppression, e.g., MIG6. More recent studies indicate that NDRG1s-inducing drugs, such as novel di-2-pyridylketone thiosemicarbazones, not only inhibit tumor growth and metastasis but also fibrous desmoplasia, which leads to chemotherapeutic resistance. The Casitas B-lineage lymphoma (c-Cbl) protein may be regulated by NDRG1, and is a crucial E3 ligase that regulates various protein tyrosine and receptor tyrosine kinases, primarily via ubiquitination. The c-Cbl protein can act as a tumor suppressor by promoting the degradation of receptor tyrosine kinases. In contrast, c-Cbl can also promote tumor development by acting as a docking protein to mediate the oncogenic c-Met/Crk/JNK and PI3K/AKT pathways. This review hypothesizes that NDRG1 could inhibit the oncogenic function of c-Cbl, which may be another mechanism of its tumor-suppressive effects.
Keywords: NDRG1; c-Cbl; pancreatic cancer; thiosemicarbazone.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
The Metastasis Suppressor, N-MYC Downstream-regulated Gene-1 (NDRG1), Down-regulates the ErbB Family of Receptors to Inhibit Downstream Oncogenic Signaling Pathways.J Biol Chem. 2016 Jan 15;291(3):1029-52. doi: 10.1074/jbc.M115.689653. Epub 2015 Nov 3. J Biol Chem. 2016. PMID: 26534963 Free PMC article.
-
The metastasis suppressor NDRG1 down-regulates the epidermal growth factor receptor via a lysosomal mechanism by up-regulating mitogen-inducible gene 6.J Biol Chem. 2019 Mar 15;294(11):4045-4064. doi: 10.1074/jbc.RA118.006279. Epub 2019 Jan 24. J Biol Chem. 2019. PMID: 30679310 Free PMC article.
-
Targeting the Metastasis Suppressor, N-Myc Downstream Regulated Gene-1, with Novel Di-2-Pyridylketone Thiosemicarbazones: Suppression of Tumor Cell Migration and Cell-Collagen Adhesion by Inhibiting Focal Adhesion Kinase/Paxillin Signaling.Mol Pharmacol. 2016 May;89(5):521-40. doi: 10.1124/mol.115.103044. Epub 2016 Feb 19. Mol Pharmacol. 2016. PMID: 26895766 Free PMC article.
-
Pharmacological targeting and the diverse functions of the metastasis suppressor, NDRG1, in cancer.Free Radic Biol Med. 2020 Sep;157:154-175. doi: 10.1016/j.freeradbiomed.2019.05.020. Epub 2019 May 24. Free Radic Biol Med. 2020. PMID: 31132412 Review.
-
Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors.Carcinogenesis. 2013 Sep;34(9):1943-54. doi: 10.1093/carcin/bgt163. Epub 2013 May 13. Carcinogenesis. 2013. PMID: 23671130 Review.
Cited by
-
Study on the Mechanism of MC5R Participating in Energy Metabolism of Goose Liver.Int J Mol Sci. 2023 May 12;24(10):8648. doi: 10.3390/ijms24108648. Int J Mol Sci. 2023. PMID: 37239994 Free PMC article.
-
A Novel Nomogram for the Preoperative Prediction of Edmondson-Steiner Grade III-IV in Hepatocellular Carcinoma Patients.J Hepatocell Carcinoma. 2023 Aug 23;10:1399-1409. doi: 10.2147/JHC.S417878. eCollection 2023. J Hepatocell Carcinoma. 2023. PMID: 37641593 Free PMC article.
-
Targeting iron to contrast cancer progression.Curr Opin Chem Biol. 2023 Jun;74:102315. doi: 10.1016/j.cbpa.2023.102315. Epub 2023 May 13. Curr Opin Chem Biol. 2023. PMID: 37187095 Free PMC article. Review.
-
Construction of a novel immune response prediction signature to predict the efficacy of immune checkpoint inhibitors in clear cell renal cell carcinoma patients.Heliyon. 2023 May 25;9(6):e15925. doi: 10.1016/j.heliyon.2023.e15925. eCollection 2023 Jun. Heliyon. 2023. PMID: 37484396 Free PMC article.
-
Re-Shaping the Pancreatic Cancer Tumor Microenvironment: A New Role for the Metastasis Suppressor NDRG1.Cancers (Basel). 2023 May 16;15(10):2779. doi: 10.3390/cancers15102779. Cancers (Basel). 2023. PMID: 37345116 Free PMC article. Review.
References
-
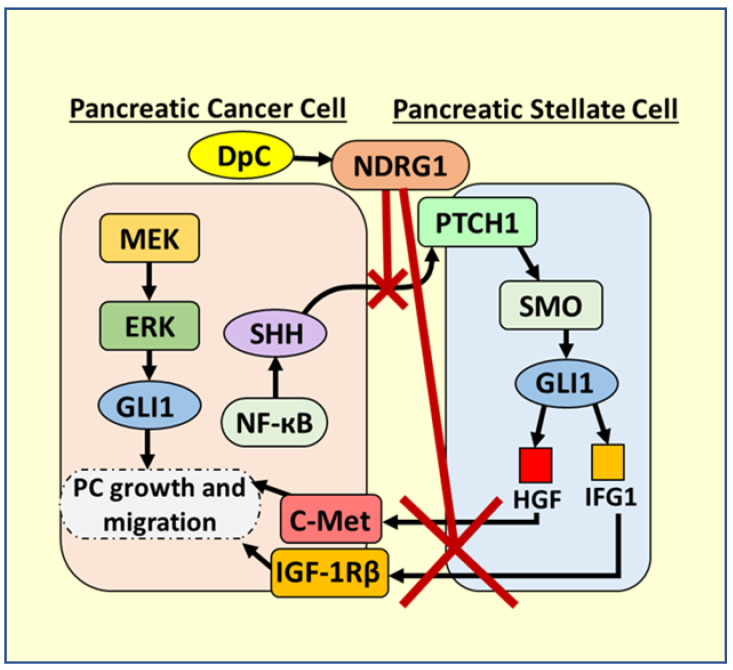
- Geleta B., Park K.C., Jansson P.J., Sahni S., Maleki S., Xu Z., Murakami T., Pajic M., Apte M.V., Richardson D.R., et al. Breaking the cycle: Targeting of NDRG1 to inhibit bi-directional oncogenic cross-talk between pancreatic cancer and stroma. FASEB J. 2021;35:e21347. doi: 10.1096/fj.202002279R. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
