

Inflammation during the life cycle of the atherosclerotic plaque
- PMID: 34550337
- PMCID: PMC8783385
- DOI: 10.1093/cvr/cvab303
Inflammation during the life cycle of the atherosclerotic plaque
Abstract
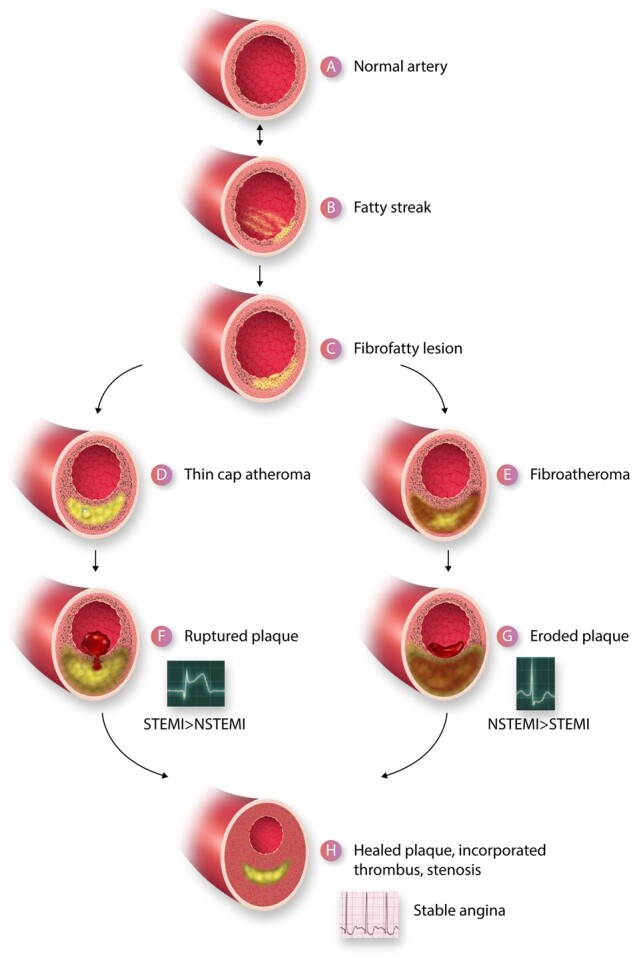
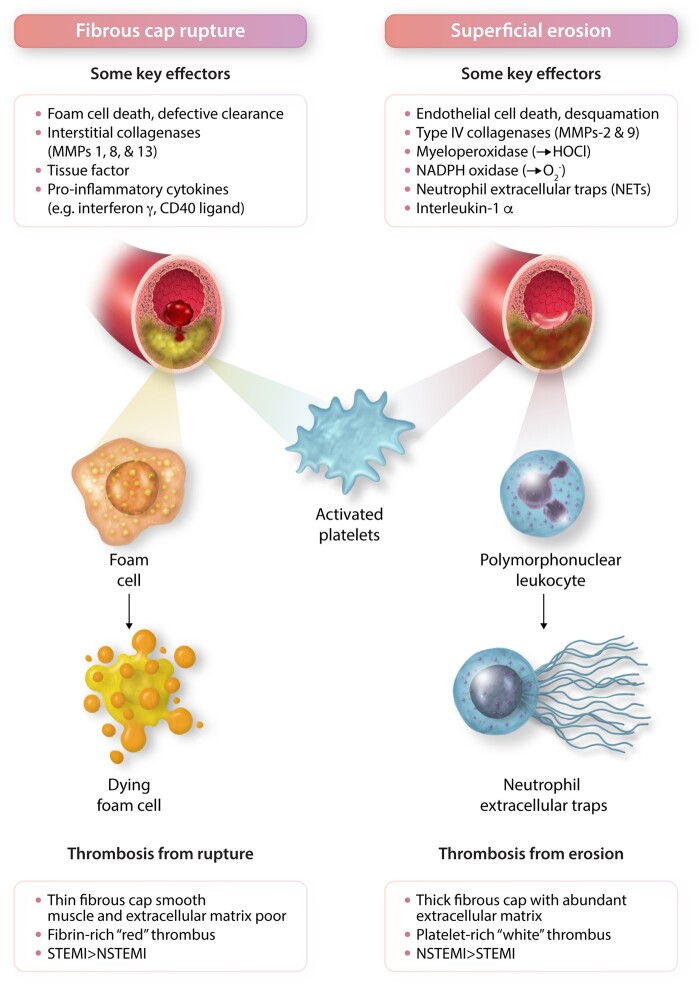
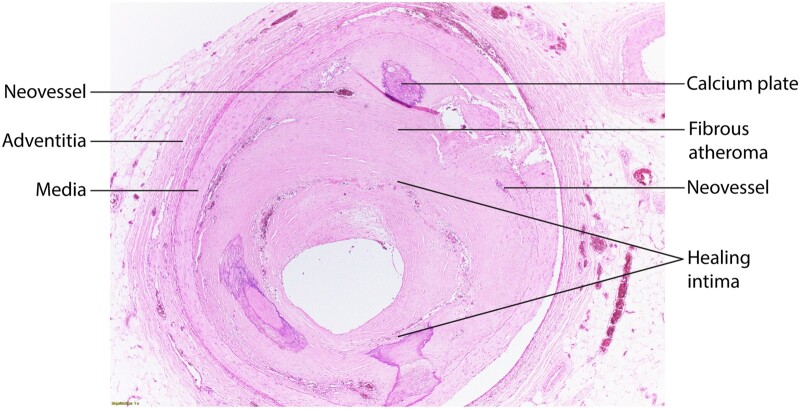
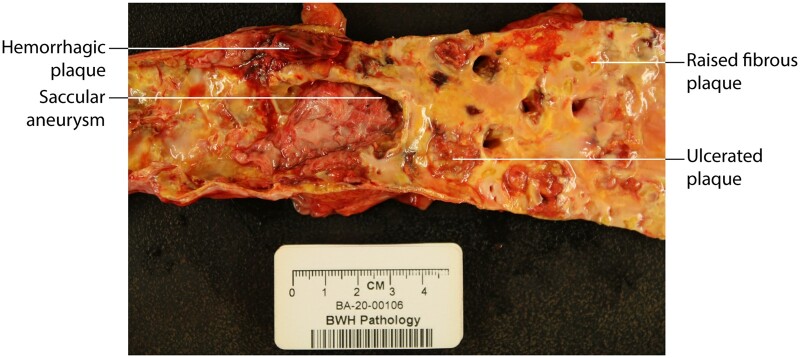
Inflammation orchestrates each stage of the life cycle of atherosclerotic plaques. Indeed, inflammatory mediators likely link many traditional and emerging risk factors with atherogenesis. Atheroma initiation involves endothelial activation with recruitment of leucocytes to the arterial intima, where they interact with lipoproteins or their derivatives that have accumulated in this layer. The prolonged and usually clinically silent progression of atherosclerosis involves periods of smouldering inflammation, punctuated by episodes of acute activation that may arise from inflammatory mediators released from sites of extravascular injury or infection or from subclinical disruptions of the plaque. Smooth muscle cells and infiltrating leucocytes can proliferate but also undergo various forms of cell death that typically lead to formation of a lipid-rich 'necrotic' core within the evolving intimal lesion. Extracellular matrix synthesized by smooth muscle cells can form a fibrous cap that overlies the lesion's core. Thus, during progression of atheroma, cells not only procreate but perish. Inflammatory mediators participate in both processes. The ultimate clinical complication of atherosclerotic plaques involves disruption that provokes thrombosis, either by fracture of the plaque's fibrous cap or superficial erosion. The consequent clots can cause acute ischaemic syndromes if they embarrass perfusion. Incorporation of the thrombi can promote plaque healing and progressive intimal thickening that can aggravate stenosis and further limit downstream blood flow. Inflammatory mediators regulate many aspects of both plaque disruption and healing process. Thus, inflammatory processes contribute to all phases of the life cycle of atherosclerotic plaques, and represent ripe targets for mitigating the disease.
Keywords: Atherosclerosis; Coronary artery disease; Endothelium; Leucocytes; Lipids; Macrophages.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author(s) 2021. For permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques.Can J Physiol Pharmacol. 2017 Oct;95(10):1245-1253. doi: 10.1139/cjpp-2016-0664. Epub 2017 Jul 26. Can J Physiol Pharmacol. 2017. PMID: 28746820 Review.
-
Macrophages and dendritic cells: the usual suspects in atherogenesis.Curr Drug Targets. 2015;16(4):373-82. doi: 10.2174/1389450116666150330115809. Curr Drug Targets. 2015. PMID: 25808566 Review.
-
Changing concepts of atherogenesis.J Intern Med. 2000 Mar;247(3):349-58. doi: 10.1046/j.1365-2796.2000.00654.x. J Intern Med. 2000. PMID: 10762452 Review.
-
2016 Russell Ross Memorial Lecture in Vascular Biology: Molecular-Cellular Mechanisms in the Progression of Atherosclerosis.Arterioscler Thromb Vasc Biol. 2017 Feb;37(2):183-189. doi: 10.1161/ATVBAHA.116.308036. Epub 2016 Dec 15. Arterioscler Thromb Vasc Biol. 2017. PMID: 27979856 Free PMC article. Review.
-
Macrophages and atherosclerotic plaque stability.Curr Opin Lipidol. 1996 Oct;7(5):330-5. doi: 10.1097/00041433-199610000-00012. Curr Opin Lipidol. 1996. PMID: 8937525 Review.
Cited by
-
Analysis of atherosclerotic plaque distribution in the carotid artery.Clin Cardiol. 2022 Dec;45(12):1272-1276. doi: 10.1002/clc.23903. Epub 2022 Sep 10. Clin Cardiol. 2022. PMID: 36086944 Free PMC article.
-
An Increasing Triglyceride-Glucose Index Is Associated with a Pro-Inflammatory and Pro-Oxidant Phenotype.J Clin Med. 2024 Jul 5;13(13):3941. doi: 10.3390/jcm13133941. J Clin Med. 2024. PMID: 38999506 Free PMC article.
-
Exploring the common mechanisms and biomarker ST8SIA4 of atherosclerosis and ankylosing spondylitis through bioinformatics analysis and machine learning.Front Cardiovasc Med. 2024 Jul 18;11:1421071. doi: 10.3389/fcvm.2024.1421071. eCollection 2024. Front Cardiovasc Med. 2024. PMID: 39131703 Free PMC article.
-
The Role of Inflammation in Cardiovascular Disease.Int J Mol Sci. 2022 Oct 26;23(21):12906. doi: 10.3390/ijms232112906. Int J Mol Sci. 2022. PMID: 36361701 Free PMC article. Review.
-
Artemvulactone E isolated from Artemisia vulgaris L. ameliorates lipopolysaccharide-induced inflammation in both RAW264.7 and zebrafish model.Front Pharmacol. 2024 Jul 18;15:1415352. doi: 10.3389/fphar.2024.1415352. eCollection 2024. Front Pharmacol. 2024. PMID: 39092222 Free PMC article.
References
-
- Li H, Cybulsky MI, Gimbrone MA Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine regulatable mononuclear leukocyte adhesion molecule, in rabbit endothelium. Arterioscler Thromb 1993;13:197–204. - PubMed
-
- Weninger WJ, Müller GB, Reiter C, Meng S, Rabl SU. Intimal hyperplasia of the infant parasellar carotid artery. Circ Res 1999;85:970–975. - PubMed
-
- Nakashima Y, Chen Y-X, Kinukawa N, Sueishi K. Distributions of diffuse intimal thickening in human arteries: preferential expression in atherosclerosis-prone arteries from an early age. Virchows Archiv 2002;441:279–288. - PubMed
-
- Schwartz SM, deBlois D, O'Brien ER. The intima: soil for atherosclerosis and restenosis. Circ Res 1995;77:445–465. - PubMed
-
- Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264–1272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
