

INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors
- PMID: 34544752
- PMCID: PMC8831444
- DOI: 10.1158/2159-8290.CD-20-1726
INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors
Abstract
Cyclin-dependent kinases 4 and 6 (CDK4/6) represent a major therapeutic vulnerability for breast cancer. The kinases are clinically targeted via ATP competitive inhibitors (CDK4/6i); however, drug resistance commonly emerges over time. To understand CDK4/6i resistance, we surveyed over 1,300 breast cancers and identified several genetic alterations (e.g., FAT1, PTEN, or ARID1A loss) converging on upregulation of CDK6. Mechanistically, we demonstrate CDK6 causes resistance by inducing and binding CDK inhibitor INK4 proteins (e.g., p18INK4C). In vitro binding and kinase assays together with physical modeling reveal that the p18INK4C-cyclin D-CDK6 complex occludes CDK4/6i binding while only weakly suppressing ATP binding. Suppression of INK4 expression or its binding to CDK6 restores CDK4/6i sensitivity. To overcome this constraint, we developed bifunctional degraders conjugating palbociclib with E3 ligands. Two resulting lead compounds potently degraded CDK4/6, leading to substantial antitumor effects in vivo, demonstrating the promising therapeutic potential for retargeting CDK4/6 despite CDK4/6i resistance. SIGNIFICANCE: CDK4/6 kinase activation represents a common mechanism by which oncogenic signaling induces proliferation and is potentially targetable by ATP competitive inhibitors. We identify a CDK6-INK4 complex that is resilient to current-generation inhibitors and develop a new strategy for more effective inhibition of CDK4/6 kinases.This article is highlighted in the In This Issue feature, p. 275.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
![Figure 1. INK4–CDK6 complex promotes resistance to CDK4/6i in cells. A, Schematic for analysis of CDK4 and CDK6 interactions and activity via coimmunoprecipitation (co-IP) followed by ADP-Glo kinase assays and mass spectrometry. B, ADP-Glo kinase assay showing immunoprecipitated CDK4 and CDK6 (IP-CDK4 and IP-CDK6) kinase activity from MCF7 parental and CDK6-high cells [cells with FAT1 CRISPR knockout (CR) that have high CDK6 expression, previously shown to have resistance to CDK4/6i; ref. 8], with or without 100 nmol/L abemaciclib treatment. Data are shown as mean + SD of three biologically independent samples. P values were determined by unpaired two-sided Student t test. RLU, relative luminescence units. C, Venn diagram showing the number of unique proteins identified by mass spectrometry coimmunoprecipitated from IP-CDK4 and IP-CDK6 in FAT1-loss cells. Percentages were calculated by number of proteins identified in each subgroup divided by total proteins identified by IP of either CDK4 or CDK6. Data are shown as means of three replicates. D, Pathway analysis by Gene Ontology of proteins interacting with CDK6 but not CDK4 in the FAT1-loss cells. The proteins were grouped by their putative biological functions. E, Unique peptide counts of cyclin-dependent kinases and their endogenous inhibitor proteins identified in the co-IP/mass spectrometry associated with CDK4 or CDK6 in the FAT1-loss cells. N = 2. F, Co-IP and immunoblotting reveal association of p15INK4B and p18INK4C with CDK6, but not CDK4, in CDK6-high cells. G, Cell line screening results showing that models with high CDKN2A or low RB1 mRNA expression are correlated with poor response to palbociclib. H, Interface residues in CDK6 in close proximity with INK4 isoforms based on previous INK4-bound CDK6 structures in the Protein Data Bank (ref. 65; no available structure for p15INK4B). CDK6-HA was immunoprecipitated using HA beads in parental MCF7 cells and MCF7 cells expressing HA-WT-CDK6-, HA-V16D-, and R31C-mutant CDK6 (disrupted INK4–CDK6 interaction) or HA-K43M/D163N-mutant CDK6 (kinase dead), and interaction with INK4 proteins was determined by immunoblotting. I, Disruption of INK4s and CDK6 binding or impairment of CDK6 kinase activity restores the sensitivity of CDK6-overexpressing cells to CDK4/6i. Cells were treated with DMSO or 100 nmol/L abemaciclib for 24 hours prior to collection. J, Percentage of cell viability of cells overexpressing WT CDK6 or R31C- or D163N-mutant CDK6 treated with increasing concentrations of abemaciclib compared with parental cells. IC50 values were recorded on day 5 following treatment. Data are shown as mean ± SD; n = 6. K, Knockdown of p15INK4B and p18INK4C in FAT1-loss cells promotes suppression of RB phosphorylation in response to abemaciclib to a similar extent as in parental cells. Cells were collected 24 hours after 100 nmol/L abemaciclib treatment. Representative blots are shown, which were repeated independently three times. L, The growth rate of p15INK4B and p18INK4C knockout in FAT1-loss cells was inhibited by 100 nmol/L abemaciclib. The cell viability was recorded at day 14 and day 21. ****, P < 0.0001. Data are shown as mean ± SD; n = 6. See also Supplementary Fig. S1.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/7d30/9762350/28719896bc2a/356fig1.jpg)
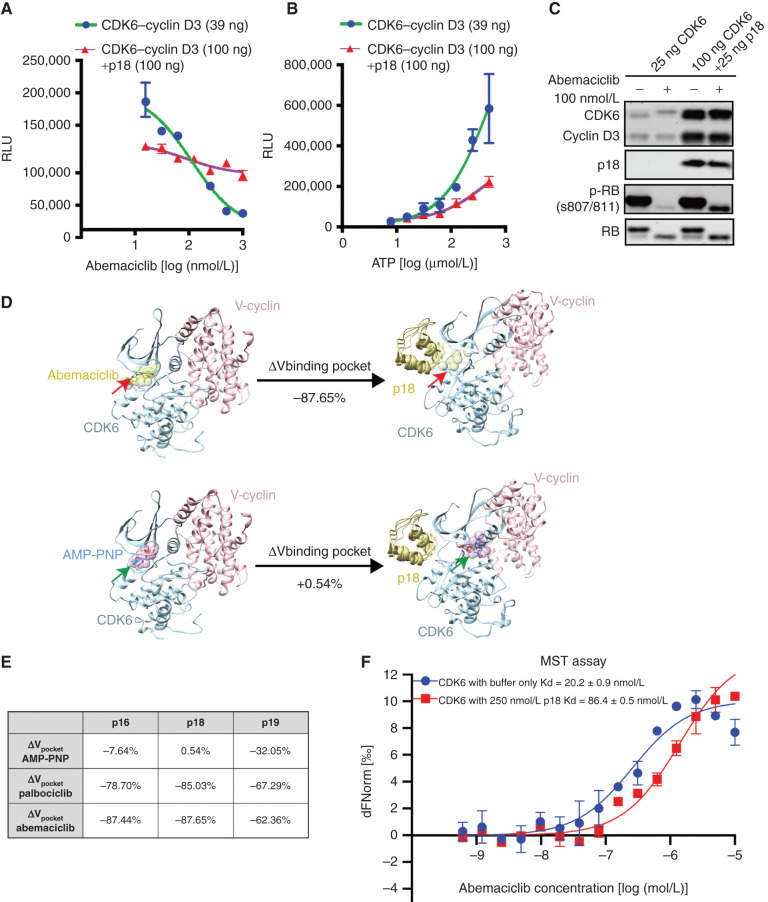
![Figure 4. Compounds targeting the CDK6–INK4 complex inhibit CDK4/6i-resistant tumors. A, Immunoblotting of MCF7 parental cells and cells with high CDK6 expression [CDK6-overexpressing (OE) cells and CDK6-high cells with FAT1 loss] treated for 24 hours with increasing concentrations of bifunctional degrader compound, BSJ-03-123, demonstrating dose-dependent targeting of CDK6 but not CDK4. B, Assessment of a panel of degrader compounds that target CDK4 and/or CDK6. Immunoblotting after 24-hour drug treatment (500 nmol/L) in FAT1-loss cells shows varying selectivity for CDK4 versus CDK6. Representative blots from three independent experiments are shown. Among them, BSJ-05-017 and BSJ-03-096 show the most significant degradation of both CDK4 and CDK6. C, Immunoblot depicting dose–response effects of BSJ-05-017 in both CDK4/6i-sensitive (left) and CDK4/6i-resistant (right) cells in comparison with palbociclib (500 nmol/L) after 24-hour treatment. D, Percentage of growth plot showing that BSJ-05-017 inhibits sensitive MCF7 parental and resistant CDK6-high cells with equal potency, whereas palbociclib shows only partial inhibition of resistant cells. IC50 values were recorded at day 7. Data are shown as mean ± SD; n = 6. E, Assay for drug-induced senescence (Senescence Green) demonstrating number of senescence marker–positive cells induced by 8 days of treatment with DMSO, BSJ-05-017 (500 nmol/L), abemaciclib (100 nmol/L), and palbociclib (500 nmol/L). BSJ-05-017 induced a significantly higher number of cells into senescence compared with abemaciclib or palbociclib in CDK6-high cells. F, Immunoblotting showing the degradation of CDK4/6 and decreased phospho-RB1 and E2F1 levels in CDK6-high (FAT1 loss) tumor-bearing mice administered 25 mg/kg BSJ-05-017 intraperitoneally. Tumors were collected 6 hours after 3 consecutive days of vehicle or BSJ-05-017 treatment (n = 2). G, Growth curve plots of cell-derived xenografts of MCF7 parental, CDK6-overexpressing, and PTEN-loss cells. Mice were treated with vehicle, ribociclib (25 mg/kg, orally), BSJ-05-017 (50 mg/kg, i.p.), or BSJ-03-096 (50 mg/kg, orally) daily for 25 to 35 days. Tumor volumes were recorded every 3 to 4 days. Data are shown as mean ± SD; n = 4. See also Supplementary Fig. S4.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/7d30/9762350/9d9ea24db08c/356fig4.jpg)
Comment on
- Cancer Discov. 12:275.
- Cancer Discov. 12:275.
Similar articles
-
Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway.Cancer Cell. 2018 Dec 10;34(6):893-905.e8. doi: 10.1016/j.ccell.2018.11.006. Cancer Cell. 2018. PMID: 30537512 Free PMC article.
-
Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors.Breast Cancer Res. 2019 Dec 18;21(1):146. doi: 10.1186/s13058-019-1230-0. Breast Cancer Res. 2019. PMID: 31852484 Free PMC article.
-
Mechanisms of sensitivity and resistance to CDK4/CDK6 inhibitors in hormone receptor-positive breast cancer treatment.Drug Resist Updat. 2024 Sep;76:101103. doi: 10.1016/j.drup.2024.101103. Epub 2024 Jun 25. Drug Resist Updat. 2024. PMID: 38943828 Review.
-
Resistance to cyclin-dependent kinase (CDK) 4/6 inhibitors confers cross-resistance to other CDK inhibitors but not to chemotherapeutic agents in breast cancer cells.Breast Cancer. 2021 Jan;28(1):206-215. doi: 10.1007/s12282-020-01150-8. Epub 2020 Aug 28. Breast Cancer. 2021. PMID: 32860163 Free PMC article.
-
Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs.Pharmacol Res. 2016 May;107:249-275. doi: 10.1016/j.phrs.2016.03.012. Epub 2016 Mar 16. Pharmacol Res. 2016. PMID: 26995305 Review.
Cited by
-
Age- and ethnic-driven molecular and clinical disparity of East Asian breast cancers.BMC Med. 2024 Sep 27;22(1):422. doi: 10.1186/s12916-024-03638-y. BMC Med. 2024. PMID: 39334392 Free PMC article.
-
Functional activation of the AKT-mTOR signalling axis in a real-world metastatic breast cancer cohort.Br J Cancer. 2024 Nov;131(9):1543-1554. doi: 10.1038/s41416-024-02852-y. Epub 2024 Sep 25. Br J Cancer. 2024. PMID: 39322687 Free PMC article.
-
EZH2 mutation is associated with the development of visceral metastasis by enhancing proliferation and invasion and inhibiting apoptosis in breast cancer cells.BMC Cancer. 2024 Sep 19;24(1):1166. doi: 10.1186/s12885-024-12950-y. BMC Cancer. 2024. PMID: 39300407 Free PMC article.
-
Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer.Nat Rev Clin Oncol. 2024 Oct;21(10):743-761. doi: 10.1038/s41571-024-00935-6. Epub 2024 Aug 23. Nat Rev Clin Oncol. 2024. PMID: 39179659 Review.
-
Strategy of combining CDK4/6 inhibitors with other therapies and mechanisms of resistance.Int J Clin Exp Pathol. 2024 Jul 15;17(7):189-207. doi: 10.62347/HGNI4903. eCollection 2024. Int J Clin Exp Pathol. 2024. PMID: 39114502 Free PMC article. Review.
References
-
- Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discovery 2004;3:1001–10. - PubMed
-
- Mian AA, Schüll M, Zhao Z, Oancea C, Hundertmark A, Beissert Tet al. . The gatekeeper mutation T315I confers resistance against small molecules by increasing or restoring the ABL-kinase activity accompanied by aberrant transphosphorylation of endogenous BCR, even in loss-of-function mutants of BCR/ABL. Leukemia 2009;23:1614–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
