

Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool
- PMID: 34527285
- PMCID: PMC8344591
- DOI: 10.1093/ve/veab064
Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool
Abstract
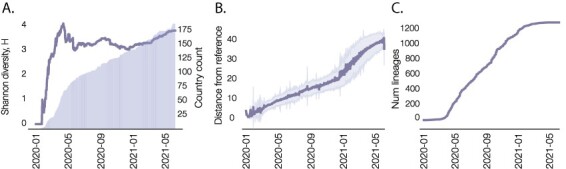
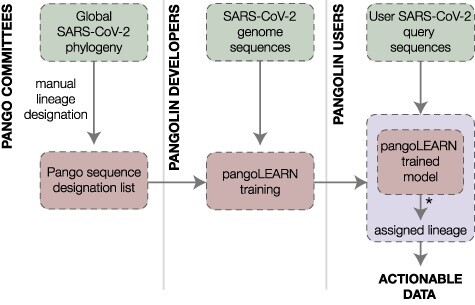
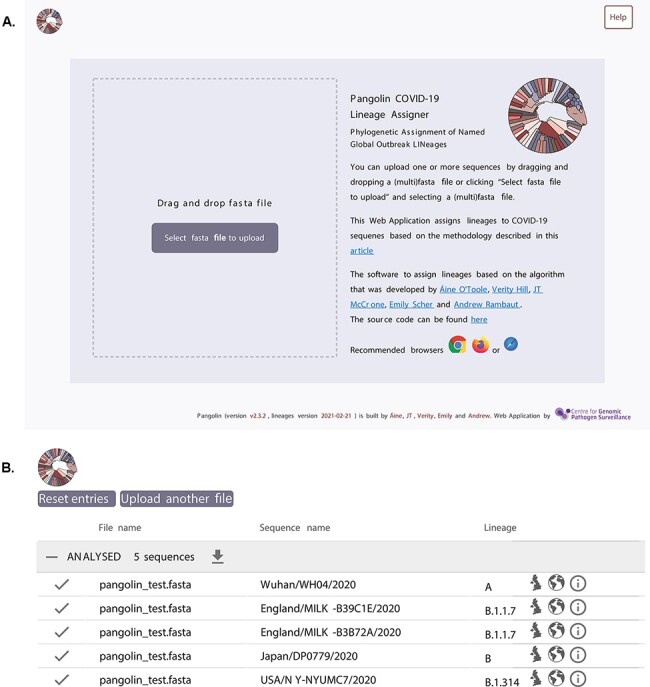
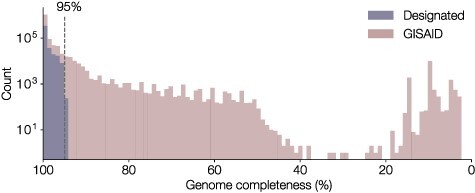
The response of the global virus genomics community to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has been unprecedented, with significant advances made towards the 'real-time' generation and sharing of SARS-CoV-2 genomic data. The rapid growth in virus genome data production has necessitated the development of new analytical methods that can deal with orders of magnitude of more genomes than previously available. Here, we present and describe Phylogenetic Assignment of Named Global Outbreak Lineages (pangolin), a computational tool that has been developed to assign the most likely lineage to a given SARS-CoV-2 genome sequence according to the Pango dynamic lineage nomenclature scheme. To date, nearly two million virus genomes have been submitted to the web-application implementation of pangolin, which has facilitated the SARS-CoV-2 genomic epidemiology and provided researchers with access to actionable information about the pandemic's transmission lineages.
Keywords: SARS-CoV-2; genomic surveillance; lineage; phylogenetics; software.
© The Author(s) 2021. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
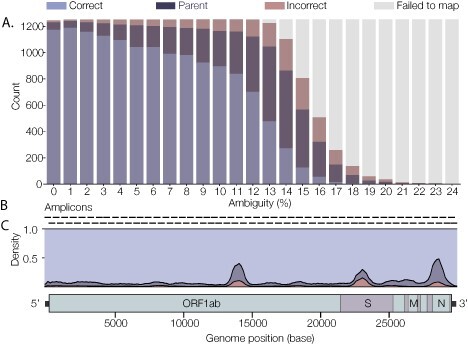

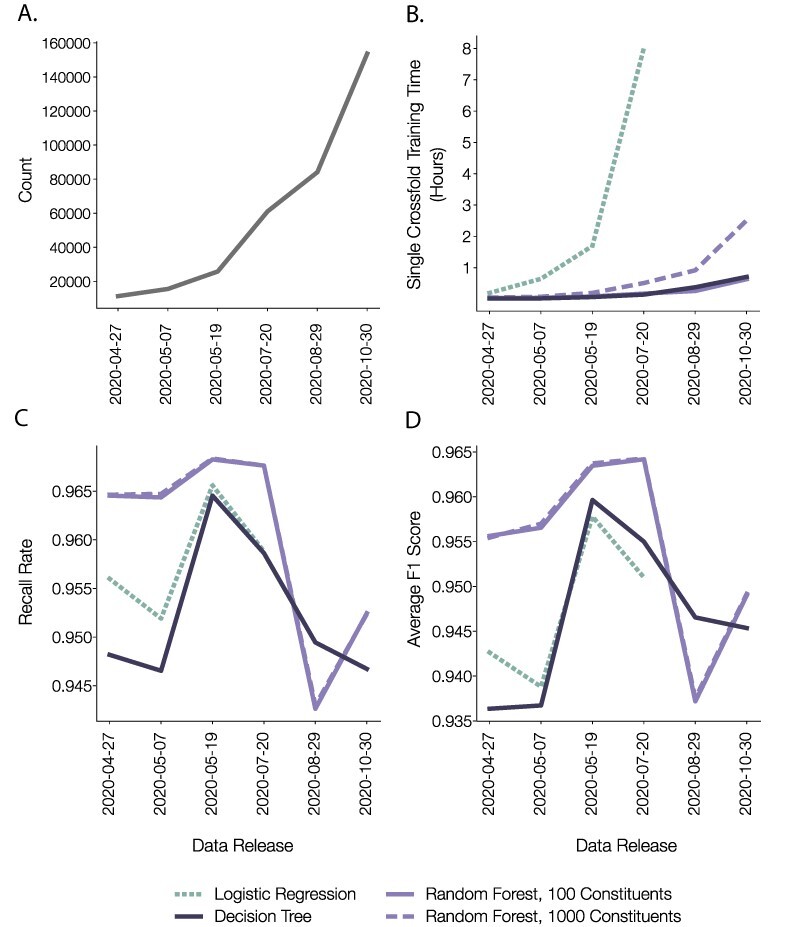
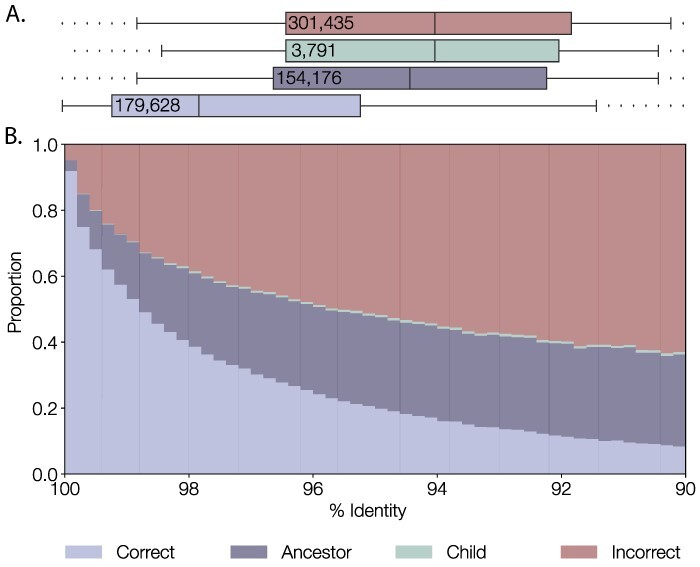
Similar articles
-
Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences.BMC Genomics. 2022 Feb 11;23(1):121. doi: 10.1186/s12864-022-08358-2. BMC Genomics. 2022. PMID: 35148677 Free PMC article.
-
Comparative genotyping of SARS-CoV-2 among Egyptian patients: near-full length genomic sequences versus selected spike and nucleocapsid regions.Med Microbiol Immunol. 2023 Dec;212(6):437-446. doi: 10.1007/s00430-023-00783-8. Epub 2023 Oct 4. Med Microbiol Immunol. 2023. PMID: 37789185 Free PMC article.
-
Vulnerability of pangolin SARS-CoV-2 lineage assignment to adversarial attack.Artif Intell Med. 2023 Dec;146:102722. doi: 10.1016/j.artmed.2023.102722. Epub 2023 Nov 18. Artif Intell Med. 2023. PMID: 38042605
-
Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations.Nat Methods. 2023 Apr;20(4):512-522. doi: 10.1038/s41592-023-01769-3. Epub 2023 Feb 23. Nat Methods. 2023. PMID: 36823332 Free PMC article.
-
Betacoronavirus Genomes: How Genomic Information has been Used to Deal with Past Outbreaks and the COVID-19 Pandemic.Int J Mol Sci. 2020 Jun 26;21(12):4546. doi: 10.3390/ijms21124546. Int J Mol Sci. 2020. PMID: 32604724 Free PMC article. Review.
Cited by
-
Recombinant SARS-CoV-2 Delta/Omicron BA.5 emerging in an immunocompromised long-term infected COVID-19 patient.Sci Rep. 2024 Oct 28;14(1):25790. doi: 10.1038/s41598-024-75241-3. Sci Rep. 2024. PMID: 39468221 Free PMC article.
-
Higher Frequency of SARS-CoV-2 RNA Shedding by Cats than Dogs in Households with Owners Recently Diagnosed with COVID-19.Viruses. 2024 Oct 11;16(10):1599. doi: 10.3390/v16101599. Viruses. 2024. PMID: 39459932 Free PMC article.
-
Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage.Viruses. 2024 Sep 26;16(10):1524. doi: 10.3390/v16101524. Viruses. 2024. PMID: 39459859 Free PMC article.
-
Emergence of the B.1.214.2 SARS-CoV-2 lineage with an Omicron-like spike insertion and a unique upper airway immune signature.BMC Infect Dis. 2024 Oct 10;24(1):1139. doi: 10.1186/s12879-024-09967-w. BMC Infect Dis. 2024. PMID: 39390446 Free PMC article.
-
SARS-CoV-2 variant replacement constrains vaccine-specific viral diversification.Virus Evol. 2024 Sep 2;10(1):veae071. doi: 10.1093/ve/veae071. eCollection 2024. Virus Evol. 2024. PMID: 39386074 Free PMC article.
References
-
- De Maio C. et al. (2020) Issues with SARS-CoV-2 Sequencing Data Virological. <https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473> accessed27 Jan 2021.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
