

The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles
- PMID: 34445323
- PMCID: PMC8395315
- DOI: 10.3390/ijms22168618
The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles
Abstract
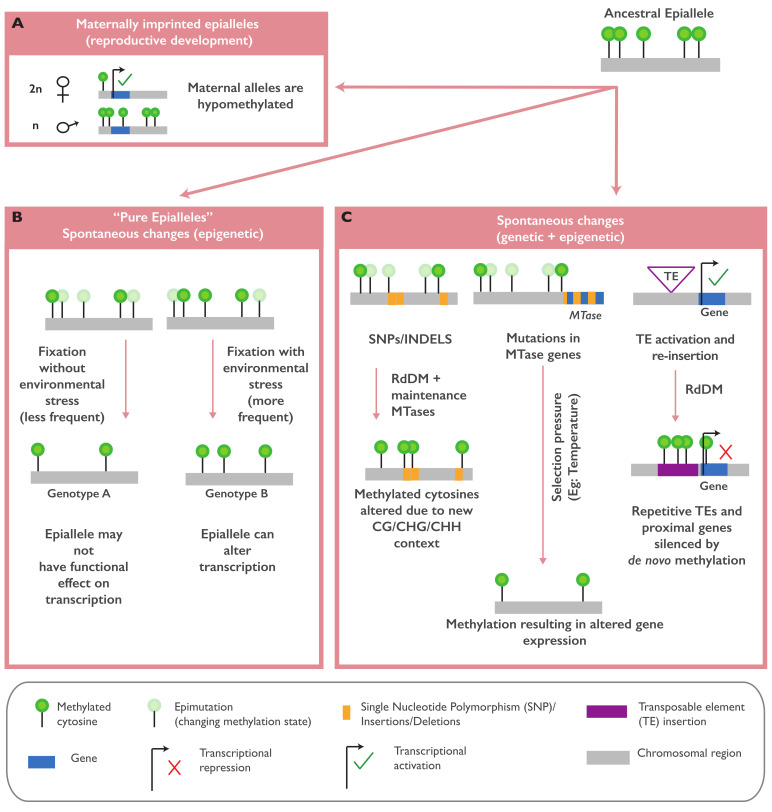
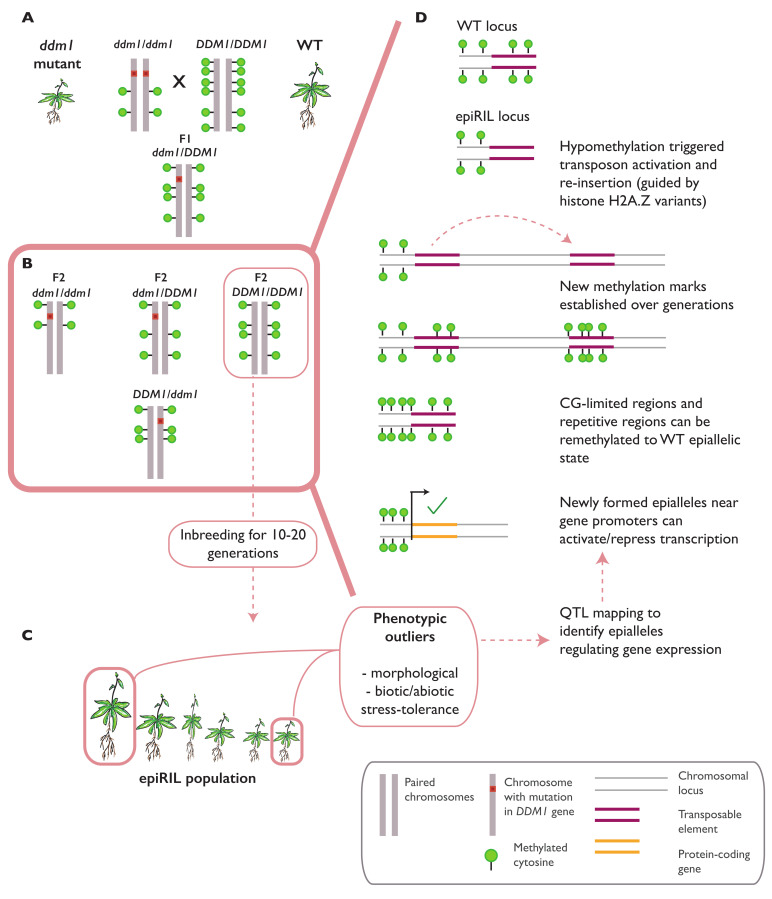
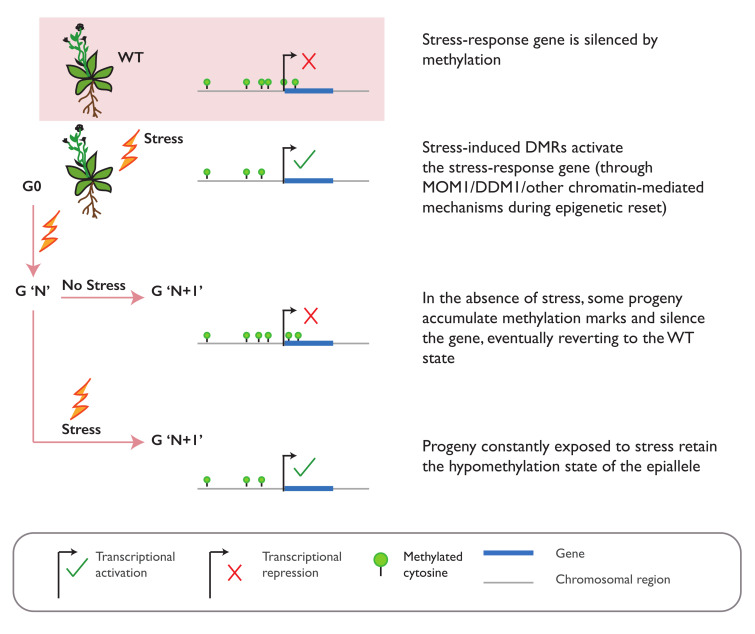
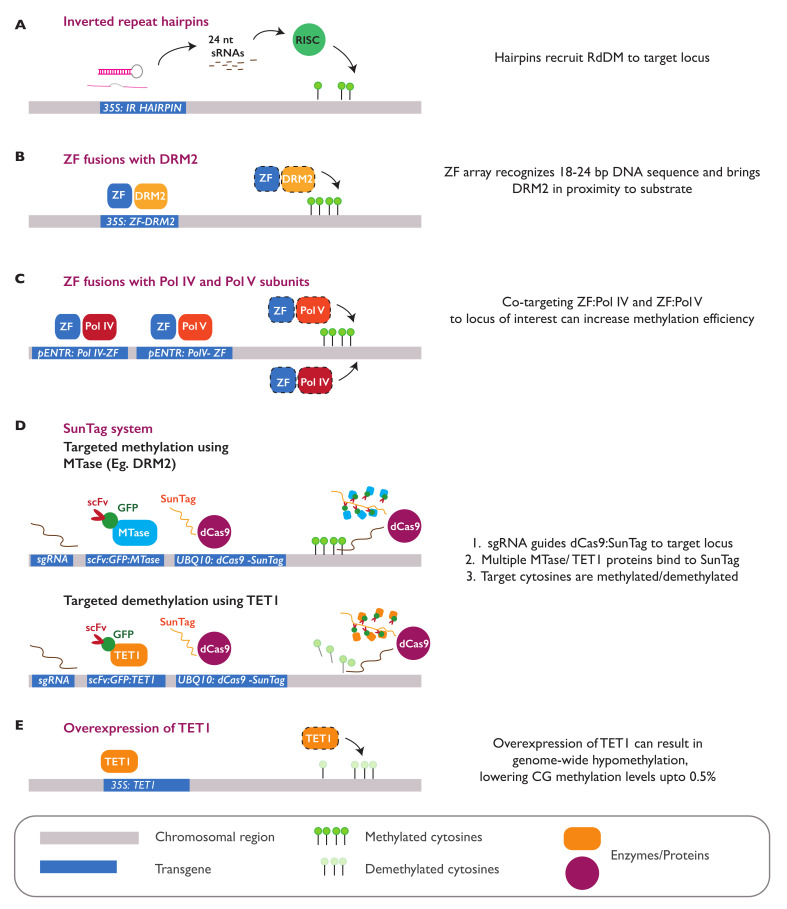
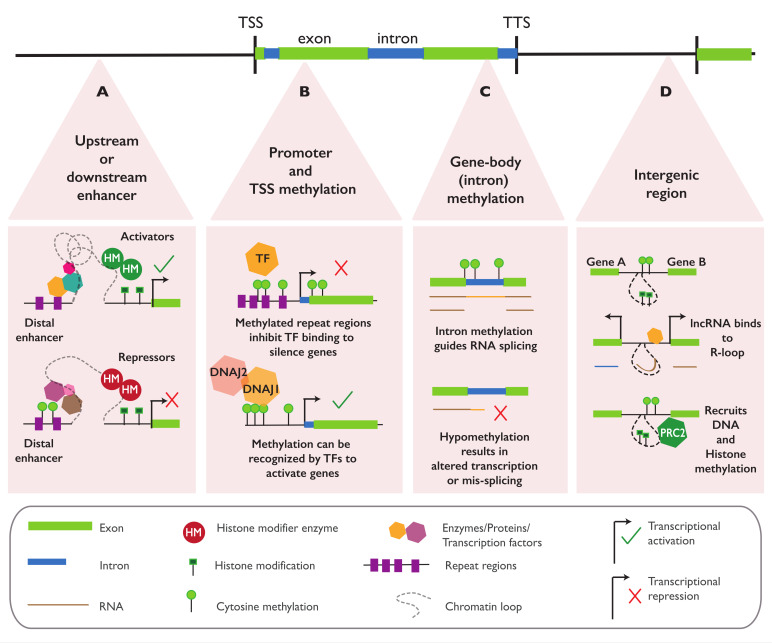
In plants, the gene expression and associated phenotypes can be modulated by dynamic changes in DNA methylation, occasionally being fixed in certain genomic loci and inherited stably as epialleles. Epiallelic variations in a population can occur as methylation changes at an individual cytosine position, methylation changes within a stretch of genomic regions, and chromatin changes in certain loci. Here, we focus on methylated regions, since it is unclear whether variations at individual methylated cytosines can serve any regulatory function, and the evidence for heritable chromatin changes independent of genetic changes is limited. While DNA methylation is known to affect and regulate wide arrays of plant phenotypes, most epialleles in the form of methylated regions have not been assigned any biological function. Here, we review how epialleles can be established in plants, serve a regulatory function, and are involved in adaptive processes. Recent studies suggest that most epialleles occur as byproducts of genetic variations, mainly from structural variants and Transposable Element (TE) activation. Nevertheless, epialleles that occur spontaneously independent of any genetic variations have also been described across different plant species. Here, we discuss how epialleles that are dependent and independent of genetic architecture are stabilized in the plant genome and how methylation can regulate a transcription relative to its genomic location.
Keywords: DNA methylation; agricultural innovation; chromatin; epiallele; gene regulation; transgenerational inheritance; transposable elements.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
-
Roles, and establishment, maintenance and erasing of the epigenetic cytosine methylation marks in plants.J Genet. 2013 Dec;92(3):629-66. doi: 10.1007/s12041-013-0273-8. J Genet. 2013. PMID: 24371187 Review.
-
The epiallelic potential of transposable elements and its evolutionary significance in plants.Philos Trans R Soc Lond B Biol Sci. 2021 Jun 7;376(1826):20200123. doi: 10.1098/rstb.2020.0123. Epub 2021 Apr 19. Philos Trans R Soc Lond B Biol Sci. 2021. PMID: 33866816 Free PMC article. Review.
-
DNA sequence properties that predict susceptibility to epiallelic switching.EMBO J. 2017 Mar 1;36(5):617-628. doi: 10.15252/embj.201695602. Epub 2017 Jan 9. EMBO J. 2017. PMID: 28069706 Free PMC article.
-
DNA cytosine methylation in plant development.J Genet Genomics. 2010 Jan;37(1):1-12. doi: 10.1016/S1673-8527(09)60020-5. J Genet Genomics. 2010. PMID: 20171573 Review.
Cited by
-
Unveiling the Mysteries of Non-Mendelian Heredity in Plant Breeding.Plants (Basel). 2023 May 11;12(10):1956. doi: 10.3390/plants12101956. Plants (Basel). 2023. PMID: 37653871 Free PMC article. Review.
-
Epigenetic Changes Occurring in Plant Inbreeding.Int J Mol Sci. 2023 Mar 12;24(6):5407. doi: 10.3390/ijms24065407. Int J Mol Sci. 2023. PMID: 36982483 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
