

RUSC2 and WDR47 oppositely regulate kinesin-1-dependent distribution of ATG9A to the cell periphery
- PMID: 34432492
- PMCID: PMC8693955
- DOI: 10.1091/mbc.E21-06-0295
RUSC2 and WDR47 oppositely regulate kinesin-1-dependent distribution of ATG9A to the cell periphery
Abstract
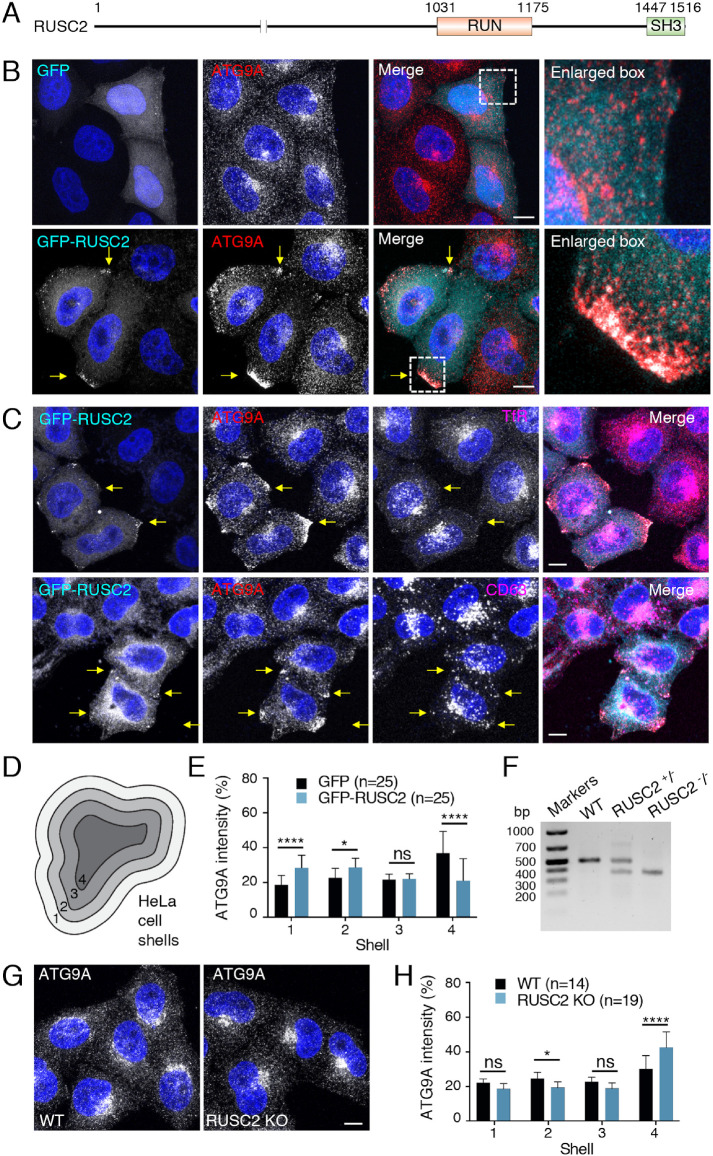
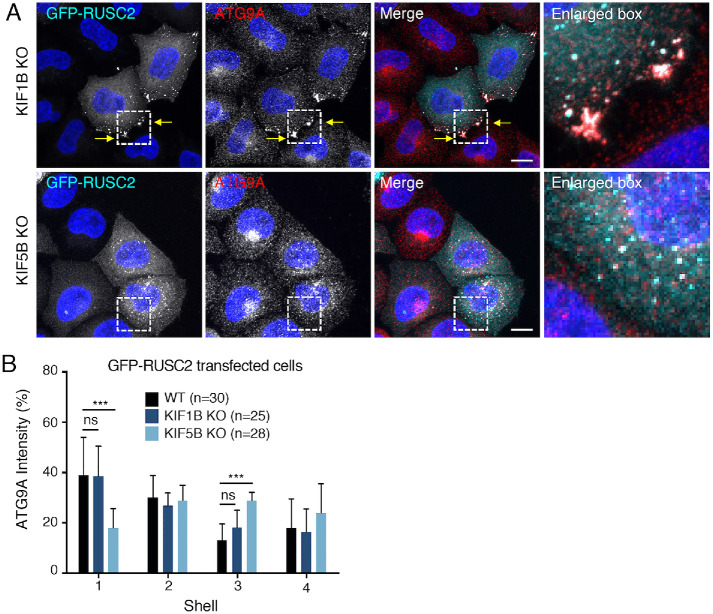
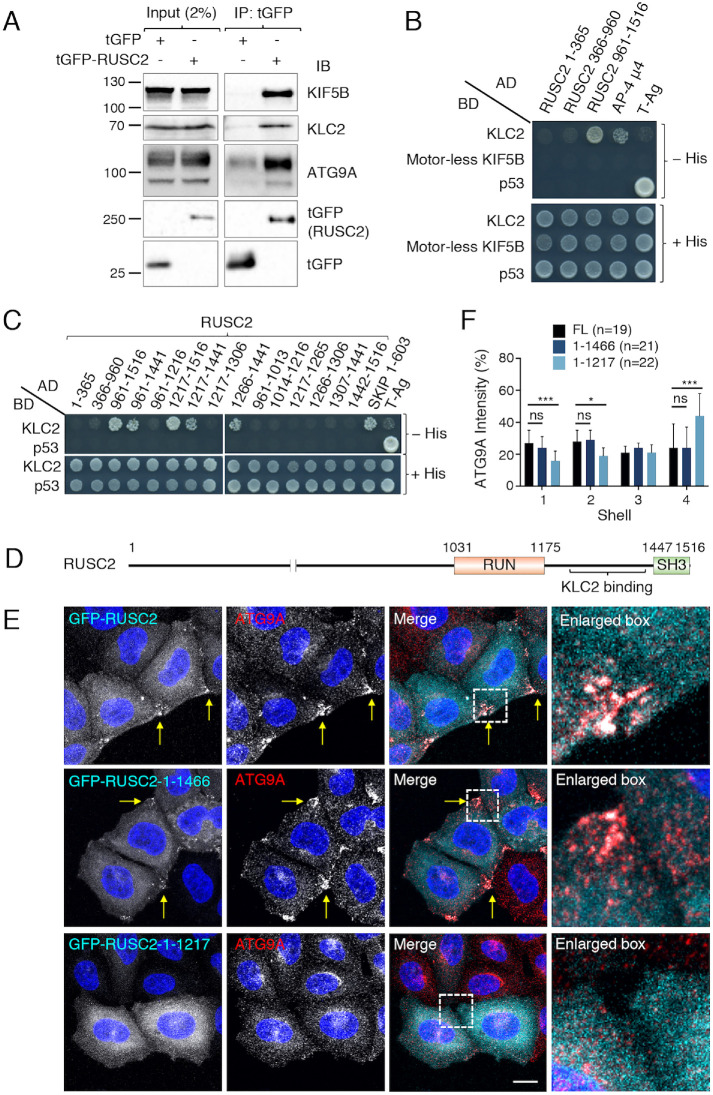
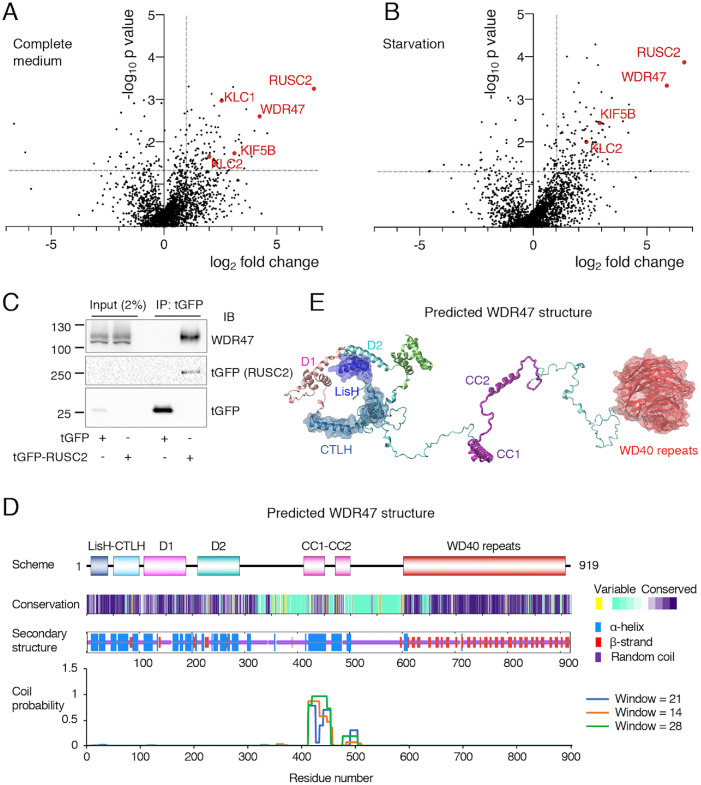
Autophagy-related protein 9 (ATG9) is a transmembrane protein component of the autophagy machinery that cycles between the trans-Golgi network (TGN) in the perinuclear area and other compartments in the peripheral area of the cell. In mammalian cells, export of the ATG9A isoform from the TGN into ATG9A-containing vesicles is mediated by the adaptor protein 4 (AP-4) complex. However, the mechanisms responsible for the subsequent distribution of these vesicles to the cell periphery are unclear. Herein we show that the AP-4-accessory protein RUSC2 couples ATG9A-containing vesicles to the plus-end-directed microtubule motor kinesin-1 via an interaction between a disordered region of RUSC2 and the kinesin-1 light chain. This interaction is counteracted by the microtubule-associated protein WDR47. These findings uncover a mechanism for the peripheral distribution of ATG9A-containing vesicles involving the function of RUSC2 as a kinesin-1 adaptor and WDR47 as a negative regulator of this function.
Figures

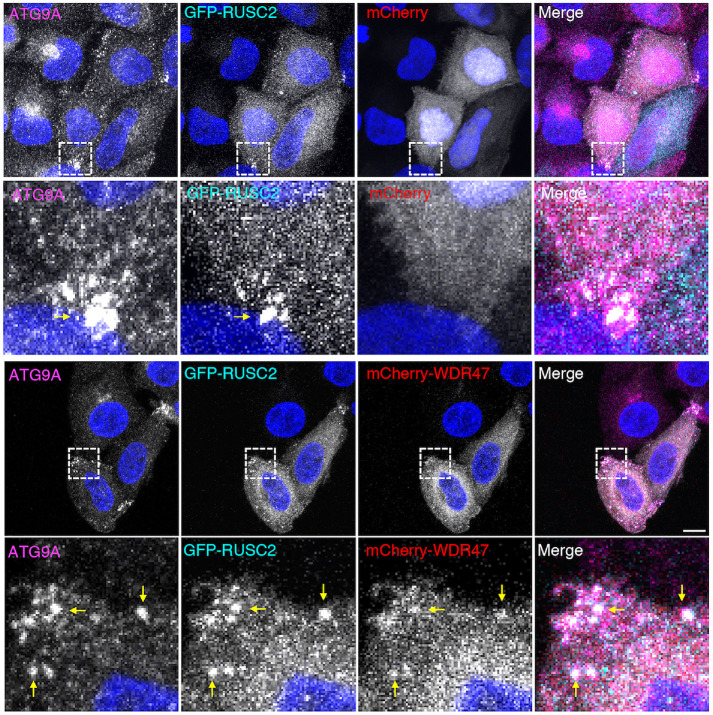
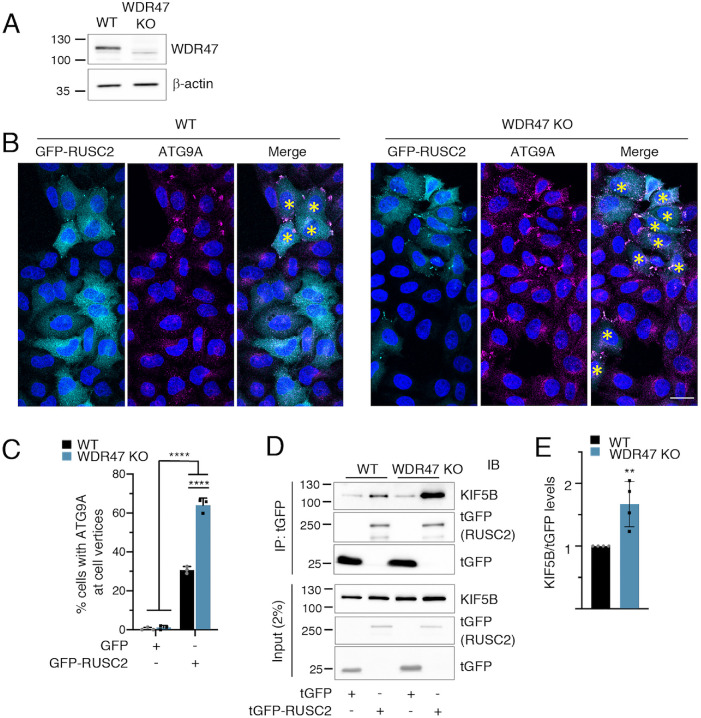
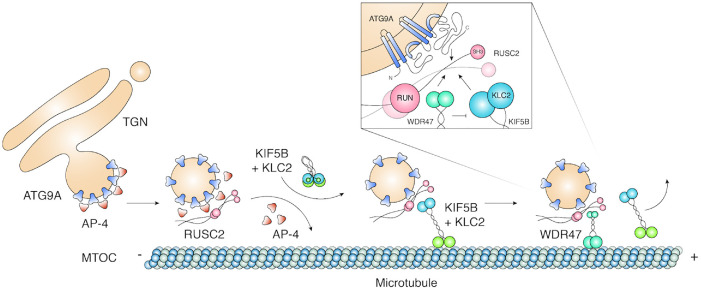
Similar articles
-
AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A.Nat Commun. 2018 Sep 27;9(1):3958. doi: 10.1038/s41467-018-06172-7. Nat Commun. 2018. PMID: 30262884 Free PMC article.
-
The FTS-Hook-FHIP (FHF) complex interacts with AP-4 to mediate perinuclear distribution of AP-4 and its cargo ATG9A.Mol Biol Cell. 2020 Apr 15;31(9):963-979. doi: 10.1091/mbc.E19-11-0658. Epub 2020 Feb 19. Mol Biol Cell. 2020. PMID: 32073997 Free PMC article.
-
The role of AP-4 in cargo export from the trans-Golgi network and hereditary spastic paraplegia.Biochem Soc Trans. 2020 Oct 30;48(5):1877-1888. doi: 10.1042/BST20190664. Biochem Soc Trans. 2020. PMID: 33084855 Review.
-
Adaptor protein complex 4 deficiency: a paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking.Hum Mol Genet. 2020 Jan 15;29(2):320-334. doi: 10.1093/hmg/ddz310. Hum Mol Genet. 2020. PMID: 31915823 Free PMC article.
-
Syntaxin 16's Newly Deciphered Roles in Autophagy.Cells. 2019 Dec 17;8(12):1655. doi: 10.3390/cells8121655. Cells. 2019. PMID: 31861136 Free PMC article. Review.
Cited by
-
Synaptic vesicle proteins and ATG9A self-organize in distinct vesicle phases within synapsin condensates.Nat Commun. 2023 Jan 28;14(1):455. doi: 10.1038/s41467-023-36081-3. Nat Commun. 2023. PMID: 36709207 Free PMC article.
-
Kinesin-1 autoinhibition facilitates the initiation of dynein cargo transport.J Cell Biol. 2023 Mar 6;222(3):e202205136. doi: 10.1083/jcb.202205136. Epub 2022 Dec 16. J Cell Biol. 2023. PMID: 36524956 Free PMC article.
-
Organization of Presynaptic Autophagy-Related Processes.Front Synaptic Neurosci. 2022 Mar 17;14:829354. doi: 10.3389/fnsyn.2022.829354. eCollection 2022. Front Synaptic Neurosci. 2022. PMID: 35368245 Free PMC article. Review.
-
Tepsin binds LC3B to promote ATG9A trafficking and delivery.Mol Biol Cell. 2024 Apr 1;35(4):ar56. doi: 10.1091/mbc.E23-09-0359-T. Epub 2024 Feb 21. Mol Biol Cell. 2024. PMID: 38381558 Free PMC article.
-
The adaptor protein chaperone AAGAB stabilizes AP-4 complex subunits.Mol Biol Cell. 2022 Oct 1;33(12):ar109. doi: 10.1091/mbc.E22-05-0177. Epub 2022 Aug 17. Mol Biol Cell. 2022. PMID: 35976721 Free PMC article.
References
-
- Abou Jamra R, Philippe O, Raas-Rothschild A, Eck SH, Graf E, Buchert R, Borck G, Ekici A, Brockschmidt FF, Nöthen MM, et al. (2011). Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet 88, 788–795. - PMC - PubMed
-
- Alwadei AH, Benini R, Mahmoud A, Alasmari A, Kamsteeg E-J, Alfadhel M (2016). Loss-of-function mutation in RUSC2 causes intellectual disability and secondary microcephaly. Dev Med Child Neurol 58, 1317–1322. - PubMed
-
- Behne R, Teinert J, Wimmer M, D’Amore A, Davies AK, Scarrott JM, Eberhardt K, Brechmann B, Chen IP, Buttermore ED, et al. (2020). Adaptor protein complex 4 deficiency: a paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking. Hum Mol Genet 29, 320–334. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
