

Application of shotgun metagenomics sequencing and targeted sequence capture to detect circulating porcine viruses in the Dutch-German border region
- PMID: 34347385
- PMCID: PMC9540031
- DOI: 10.1111/tbed.14249
Application of shotgun metagenomics sequencing and targeted sequence capture to detect circulating porcine viruses in the Dutch-German border region
Abstract
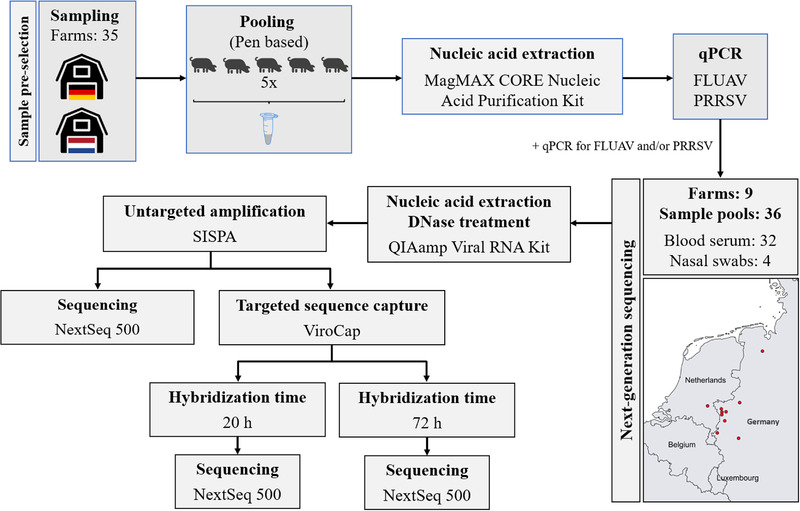
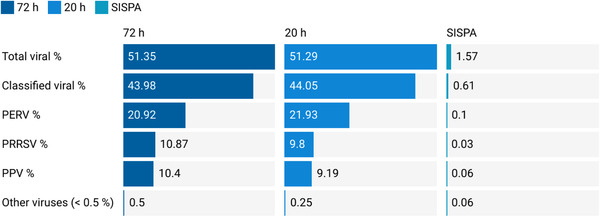
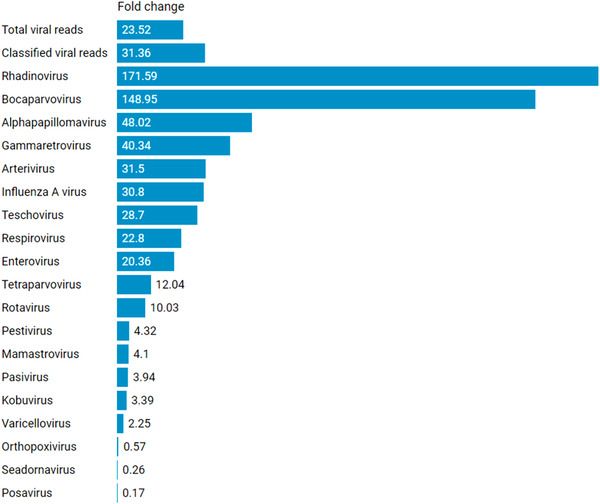
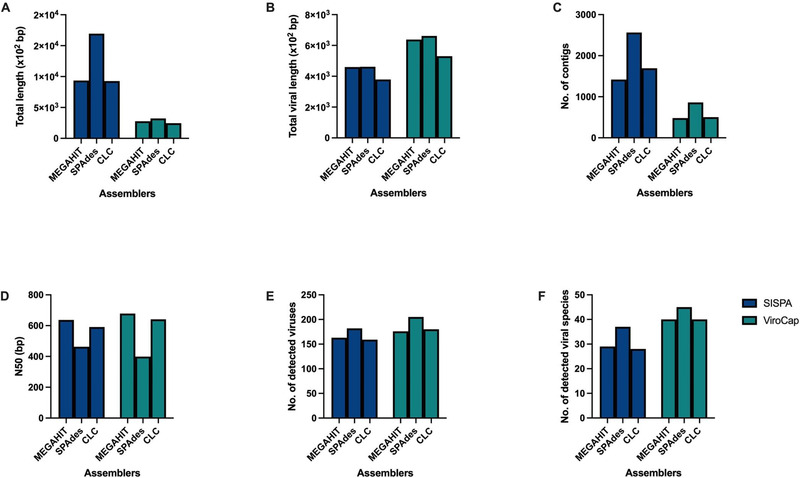
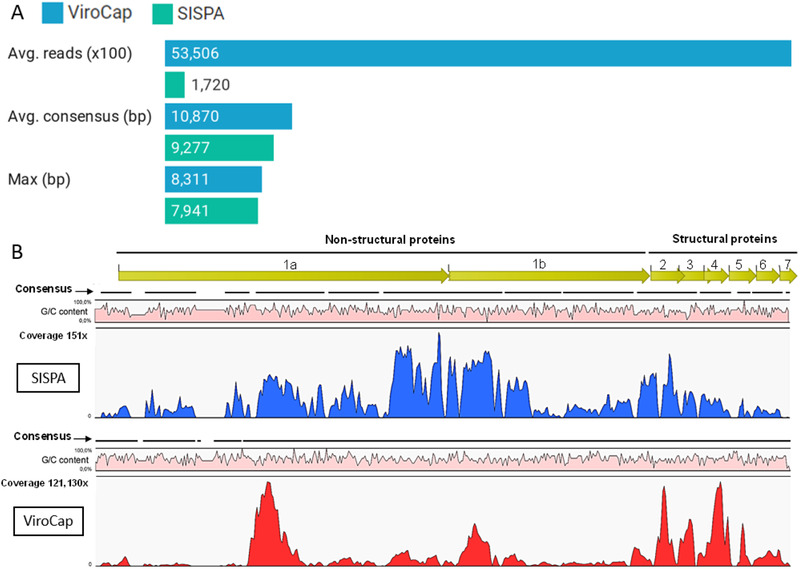
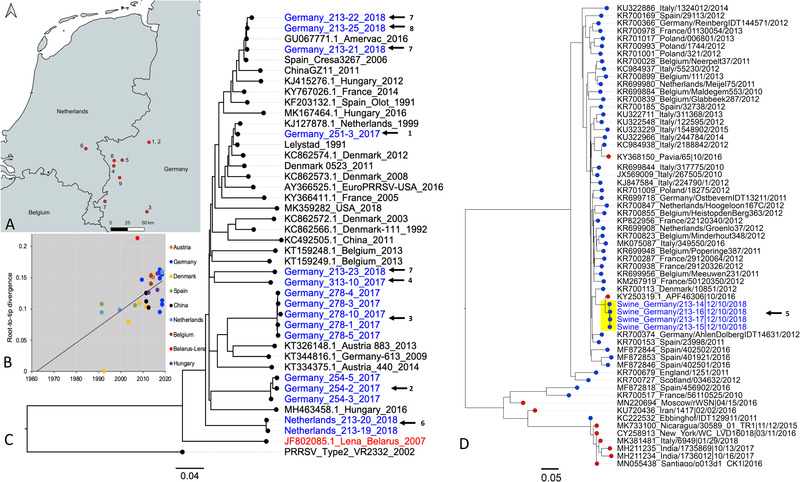
Porcine viruses have been emerging in recent decades, threatening animal and human health, as well as economic stability for pig farmers worldwide. Next-generation sequencing (NGS) can detect and characterize known and unknown viruses but has limited sensitivity when an unbiased approach, such as shotgun metagenomics sequencing, is used. To increase the sensitivity of NGS for the detection of viruses, we applied and evaluated a broad viral targeted sequence capture (TSC) panel and compared it to an unbiased shotgun metagenomic approach. A cohort of 36 pooled porcine nasal swab and blood serum samples collected from both sides of the Dutch-German border region were evaluated. Overall, we detected 46 different viral species using TSC, compared to 40 viral species with a shotgun metagenomics approach. Furthermore, we performed phylogenetic analysis on recovered influenza A virus (FLUAV) genomes from Germany and revealed a close similarity to a zoonotic influenza strain previously detected in the Netherlands. Although TSC introduced coverage bias within the detected viruses, it improved sensitivity, genome sequence depth and contig length. In-depth characterization of the swine virome, coupled with developing new enrichment techniques, can play a crucial role in the surveillance of circulating porcine viruses and emerging zoonotic pathogens.
Keywords: influenza A virus; one health; porcine virome; shotgun metagenomics sequencing; surveillance; targeted sequence capture.
© 2021 The Authors. Transboundary and Emerging Diseases published by Wiley-VCH GmbH.
Conflict of interest statement
John W. A. Rossen is employed by IDbyDNA. Silke Peter consults for IDbyDNA. This did not influence the interpretation of reviewed data and conclusions drawn nor the drafting of the manuscript, and no support was obtained from them. All other authors declare no conflict of interest.
Figures
Similar articles
-
Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples.Viruses. 2020 Nov 27;12(12):1358. doi: 10.3390/v12121358. Viruses. 2020. PMID: 33260903 Free PMC article.
-
Enhanced virome sequencing using targeted sequence capture.Genome Res. 2015 Dec;25(12):1910-20. doi: 10.1101/gr.191049.115. Epub 2015 Sep 22. Genome Res. 2015. PMID: 26395152 Free PMC article.
-
Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance.Nat Microbiol. 2020 Mar;5(3):443-454. doi: 10.1038/s41564-019-0637-9. Epub 2020 Jan 13. Nat Microbiol. 2020. PMID: 31932713 Free PMC article.
-
Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine.Viruses. 2022 Nov 4;14(11):2448. doi: 10.3390/v14112448. Viruses. 2022. PMID: 36366546 Free PMC article. Review.
-
From the Pipeline to the Bedside: Advances and Challenges in Clinical Metagenomics.J Infect Dis. 2020 Mar 28;221(Suppl 3):S331-S340. doi: 10.1093/infdis/jiz151. J Infect Dis. 2020. PMID: 31538184 Free PMC article. Review.
Cited by
-
Application of Host-Depleted Nanopore Metagenomic Sequencing in the Clinical Detection of Pathogens in Pigs and Cats.Animals (Basel). 2023 Dec 13;13(24):3838. doi: 10.3390/ani13243838. Animals (Basel). 2023. PMID: 38136875 Free PMC article.
-
Screening for Viruses in Indigenous Greek Black Pigs.Microorganisms. 2024 Feb 2;12(2):315. doi: 10.3390/microorganisms12020315. Microorganisms. 2024. PMID: 38399719 Free PMC article.
-
Non-Targeted RNA Sequencing: Towards the Development of Universal Clinical Diagnosis Methods for Human and Veterinary Infectious Diseases.Vet Sci. 2024 May 26;11(6):239. doi: 10.3390/vetsci11060239. Vet Sci. 2024. PMID: 38921986 Free PMC article. Review.
-
Swine Norovirus: Past, Present, and Future.Viruses. 2022 Mar 5;14(3):537. doi: 10.3390/v14030537. Viruses. 2022. PMID: 35336944 Free PMC article. Review.
-
The impact and complete genome characterisation of viruses involved in outbreaks of gastroenteritis in a farrow-to-finish holding.Sci Rep. 2023 Oct 31;13(1):18780. doi: 10.1038/s41598-023-45994-4. Sci Rep. 2023. PMID: 37907693 Free PMC article.
References
-
- Balka, G. , Podgórska, K. , Brar, M. S. , Bálint, Á. , Cadar, D. , Celer, V. , Dénes, L. , Dirbakova, Z. , Jedryczko, A. , Márton, L. , Novosel, D. , Petrović, T. , Sirakov, I. , Szalay, D. , Toplak, I. , Leung, F. C.‐.C. , & Stadejek, T. (2018). Genetic diversity of PRRSV 1 in Central Eastern Europe in 1994–2014: Origin and evolution of the virus in the region. Scientific Reports, 8(1), 1–12. 10.1038/s41598-018-26036-w - DOI - PMC - PubMed
-
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19(5), 455–477. 10.1089/cmb.2012.0021 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
