

Anti-tumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10
- PMID: 34297038
- PMCID: PMC8313406
- DOI: 10.1084/jem.20210108
Anti-tumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10
Abstract
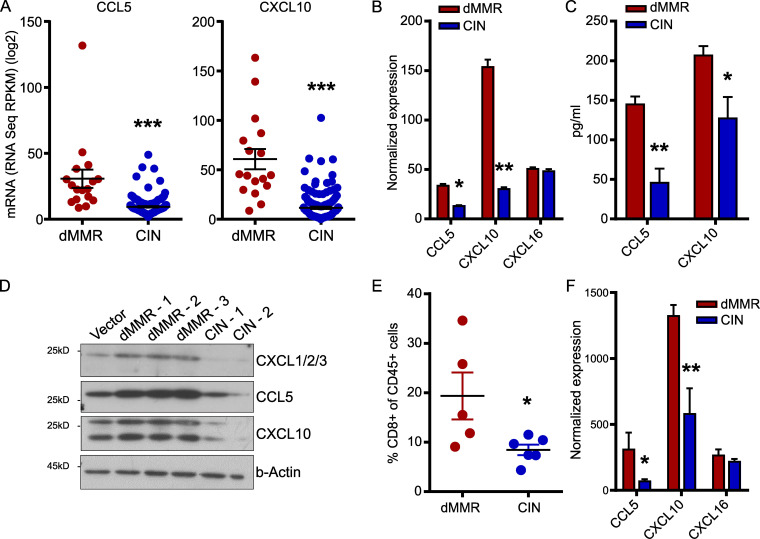
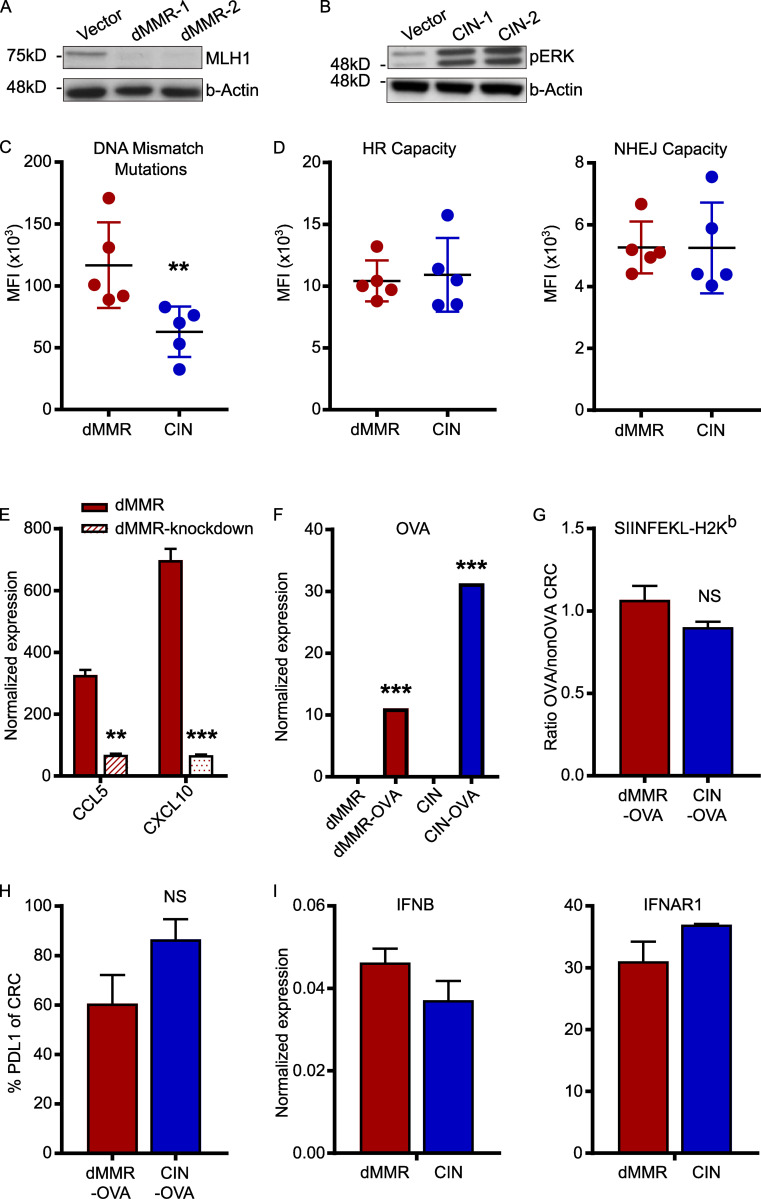
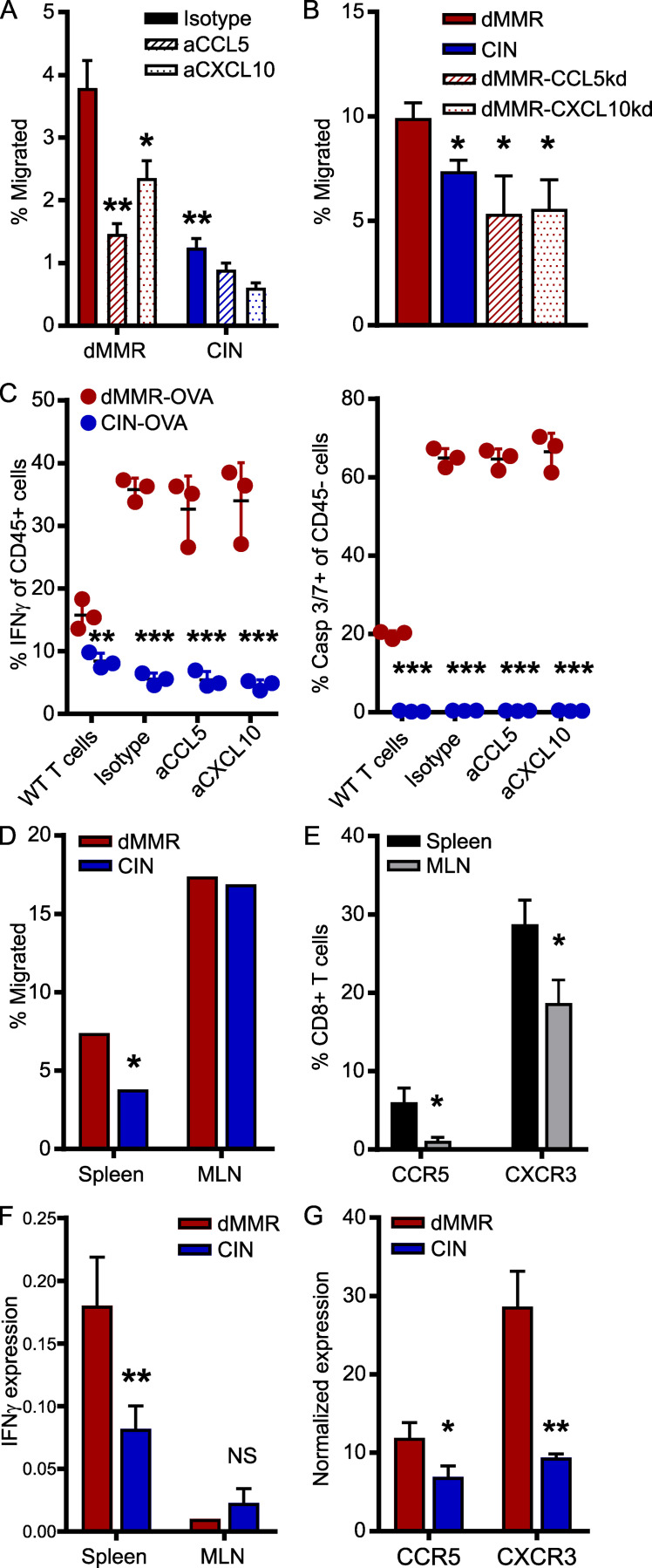
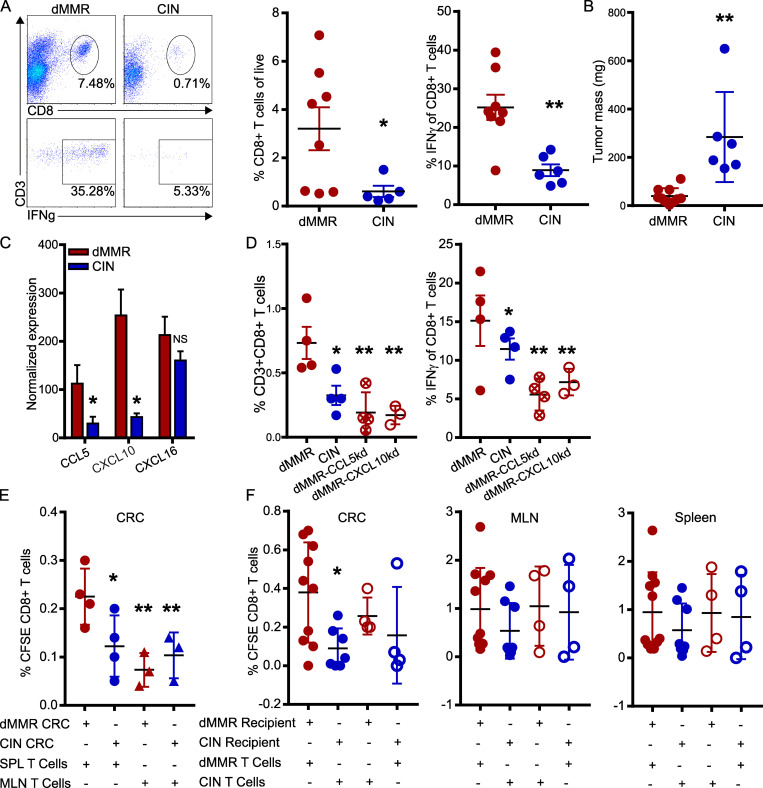
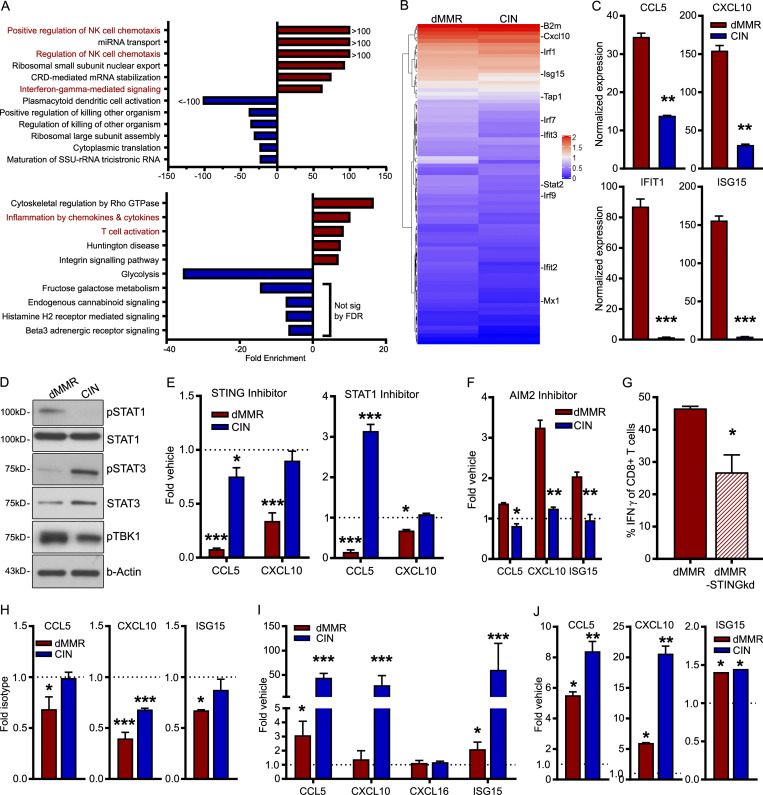
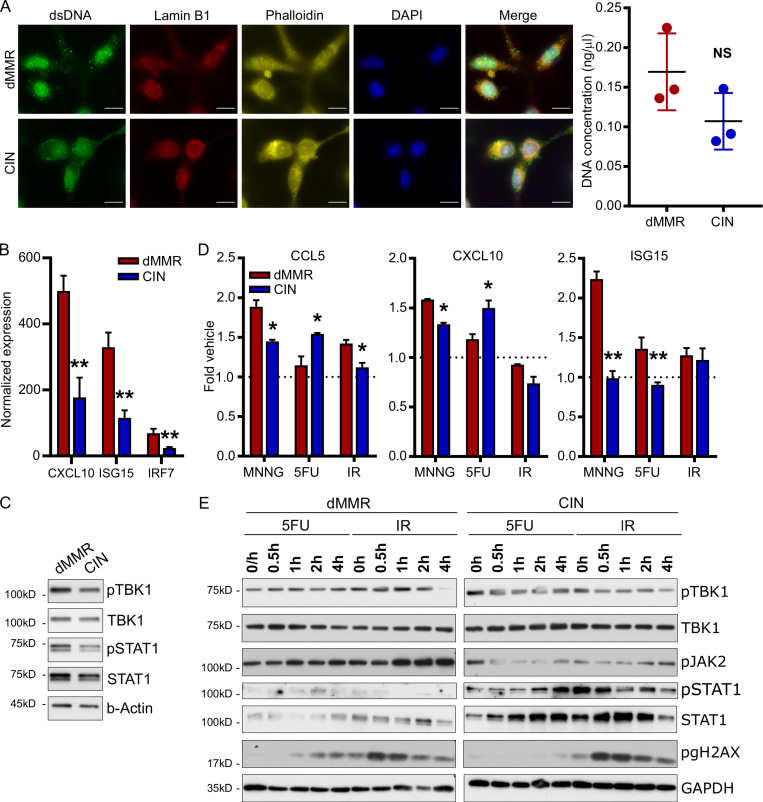
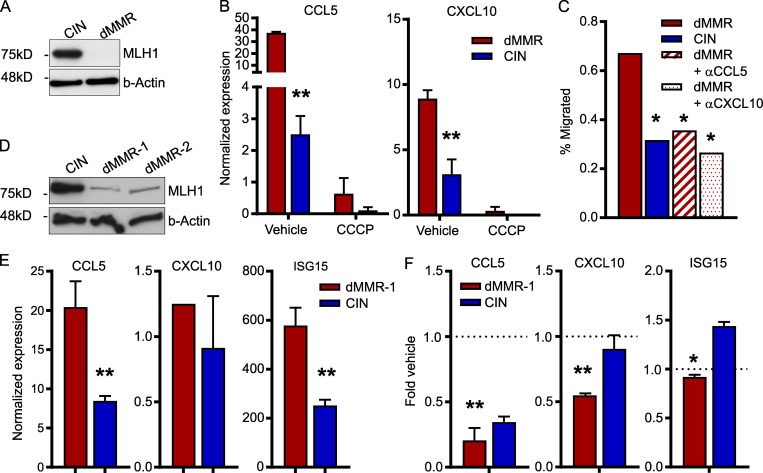
Colorectal cancers (CRCs) deficient in DNA mismatch repair (dMMR) contain abundant CD8+ tumor-infiltrating lymphocytes (TILs) responding to the abundant neoantigens from their unstable genomes. Priming of such tumor-targeted TILs first requires recruitment of CD8+ T cells into the tumors, implying that this is an essential prerequisite of successful dMMR anti-tumor immunity. We have discovered that selective recruitment and activation of systemic CD8+ T cells into dMMR CRCs strictly depend on overexpression of CCL5 and CXCL10 due to endogenous activation of cGAS/STING and type I IFN signaling by damaged DNA. TIL infiltration into orthotopic dMMR CRCs is neoantigen-independent and followed by induction of a resident memory-like phenotype key to the anti-tumor response. CCL5 and CXCL10 could be up-regulated by common chemotherapies in all CRCs, indicating that facilitating CD8+ T cell recruitment underlies their efficacy. Induction of CCL5 and CXCL10 thus represents a tractable therapeutic strategy to induce TIL recruitment into CRCs, where local priming can be maximized even in neoantigen-poor CRCs.
© 2021 Mowat et al.
Conflict of interest statement
Disclosures: K. Baker reported grants from Canadian Institutes of Health Research and from Canadian Foundation for Innovation during the conduct of the study. No other disclosures were reported.
Figures
Similar articles
-
Role of the cGAS-STING pathway in regulating the tumor-immune microenvironment in dMMR/MSI colorectal cancer.Cancer Immunol Immunother. 2022 Nov;71(11):2765-2776. doi: 10.1007/s00262-022-03200-w. Epub 2022 Apr 16. Cancer Immunol Immunother. 2022. PMID: 35429245 Free PMC article.
-
Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells.Cancer Res. 2012 Mar 1;72(5):1092-102. doi: 10.1158/0008-5472.CAN-11-2493. Epub 2012 Jan 26. Cancer Res. 2012. PMID: 22282655
-
Dickkopf 1 impairs the tumor response to PD-1 blockade by inactivating CD8+ T cells in deficient mismatch repair colorectal cancer.J Immunother Cancer. 2021 Mar;9(3):e001498. doi: 10.1136/jitc-2020-001498. J Immunother Cancer. 2021. PMID: 33782107 Free PMC article.
-
Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside.Biochim Biophys Acta Rev Cancer. 2020 Dec;1874(2):188447. doi: 10.1016/j.bbcan.2020.188447. Epub 2020 Oct 6. Biochim Biophys Acta Rev Cancer. 2020. PMID: 33035640 Free PMC article. Review.
-
Deficient Mismatch Repair and the Role of Immunotherapy in Metastatic Colorectal Cancer.Curr Treat Options Oncol. 2016 Aug;17(8):41. doi: 10.1007/s11864-016-0414-4. Curr Treat Options Oncol. 2016. PMID: 27315067 Review.
Cited by
-
Identification of Potential Prognostic and Predictive Immunological Biomarkers in Patients with Stage I and Stage III Non-Small Cell Lung Cancer (NSCLC): A Prospective Exploratory Study.Cancers (Basel). 2021 Dec 13;13(24):6259. doi: 10.3390/cancers13246259. Cancers (Basel). 2021. PMID: 34944879 Free PMC article.
-
Tumor Immunogenic Cell Death as a Mediator of Intratumor CD8 T-Cell Recruitment.Cells. 2022 Nov 18;11(22):3672. doi: 10.3390/cells11223672. Cells. 2022. PMID: 36429101 Free PMC article. Review.
-
The senescence journey in cancer immunoediting.Mol Cancer. 2024 Apr 1;23(1):68. doi: 10.1186/s12943-024-01973-5. Mol Cancer. 2024. PMID: 38561826 Free PMC article. Review.
-
Chemokine Homeostasis in Healthy Volunteers and during Pancreatic and Colorectal Tumor Growth in Murine Models.Curr Issues Mol Biol. 2022 Oct 18;44(10):4987-4999. doi: 10.3390/cimb44100339. Curr Issues Mol Biol. 2022. PMID: 36286054 Free PMC article.
-
cGAS-STING pathway as a potential trigger of immunosenescence and inflammaging.Front Immunol. 2023 Feb 28;14:1132653. doi: 10.3389/fimmu.2023.1132653. eCollection 2023. Front Immunol. 2023. PMID: 36926349 Free PMC article. Review.
References
-
- Baker, K., Zlobec I., Tornillo L., Terracciano L., Jass J.R., and Lugli A.. 2007. Differential significance of tumour infiltrating lymphocytes in sporadic mismatch repair deficient versus proficient colorectal cancers: a potential role for dysregulation of the transforming growth factor-beta pathway. Eur. J. Cancer. 43:624–631. 10.1016/j.ejca.2006.11.012 - DOI - PubMed
-
- Baker, K., Qiao S.W., Kuo T.T., Aveson V.G., Platzer B., Andersen J.T., Sandlie I., Chen Z., de Haar C., Lencer W.I., et al. . 2011. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. USA. 108:9927–9932. 10.1073/pnas.1019037108 - DOI - PMC - PubMed
-
- Baker, K., Rath T., Flak M.B., Arthur J.C., Chen Z., Glickman J.N., Zlobec I., Karamitopoulou E., Stachler M.D., Odze R.D., et al. . 2013. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity. 39:1095–1107. 10.1016/j.immuni.2013.11.003 - DOI - PMC - PubMed
-
- Bindea, G., Mlecnik B., Tosolini M., Kirilovsky A., Waldner M., Obenauf A.C., Angell H., Fredriksen T., Lafontaine L., Berger A., et al. . 2013. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 39:782–795. 10.1016/j.immuni.2013.10.003 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
