

Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation
- PMID: 34296965
- PMCID: PMC8461839
- DOI: 10.1152/ajpheart.00125.2021
Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation
Abstract
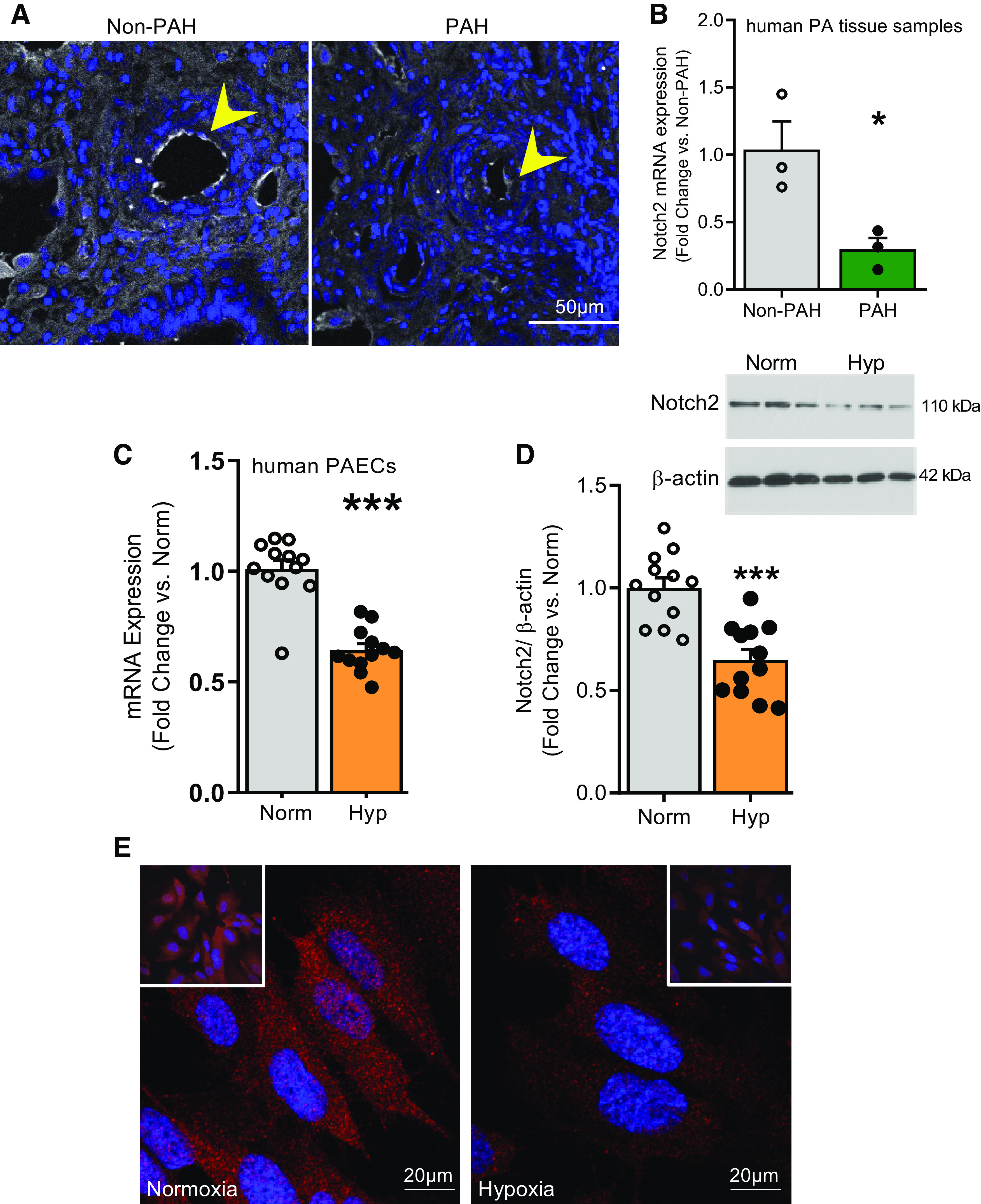
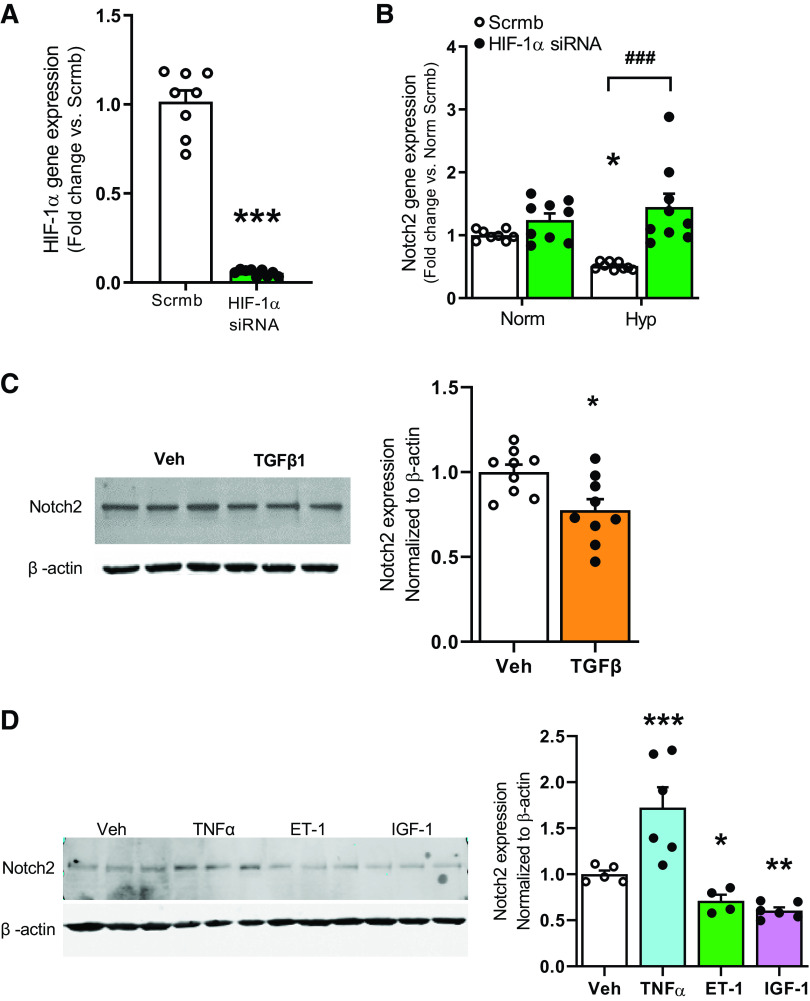
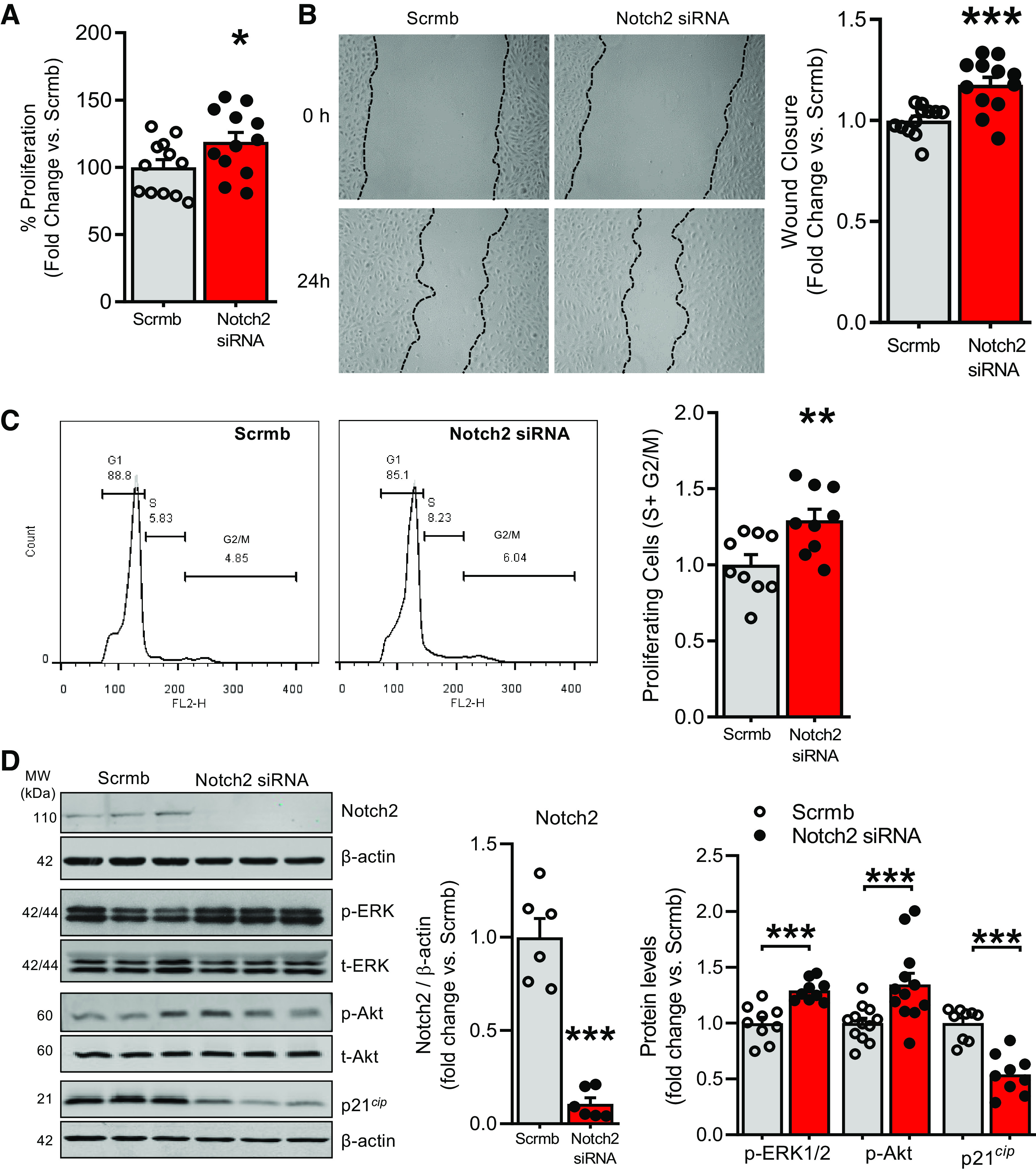
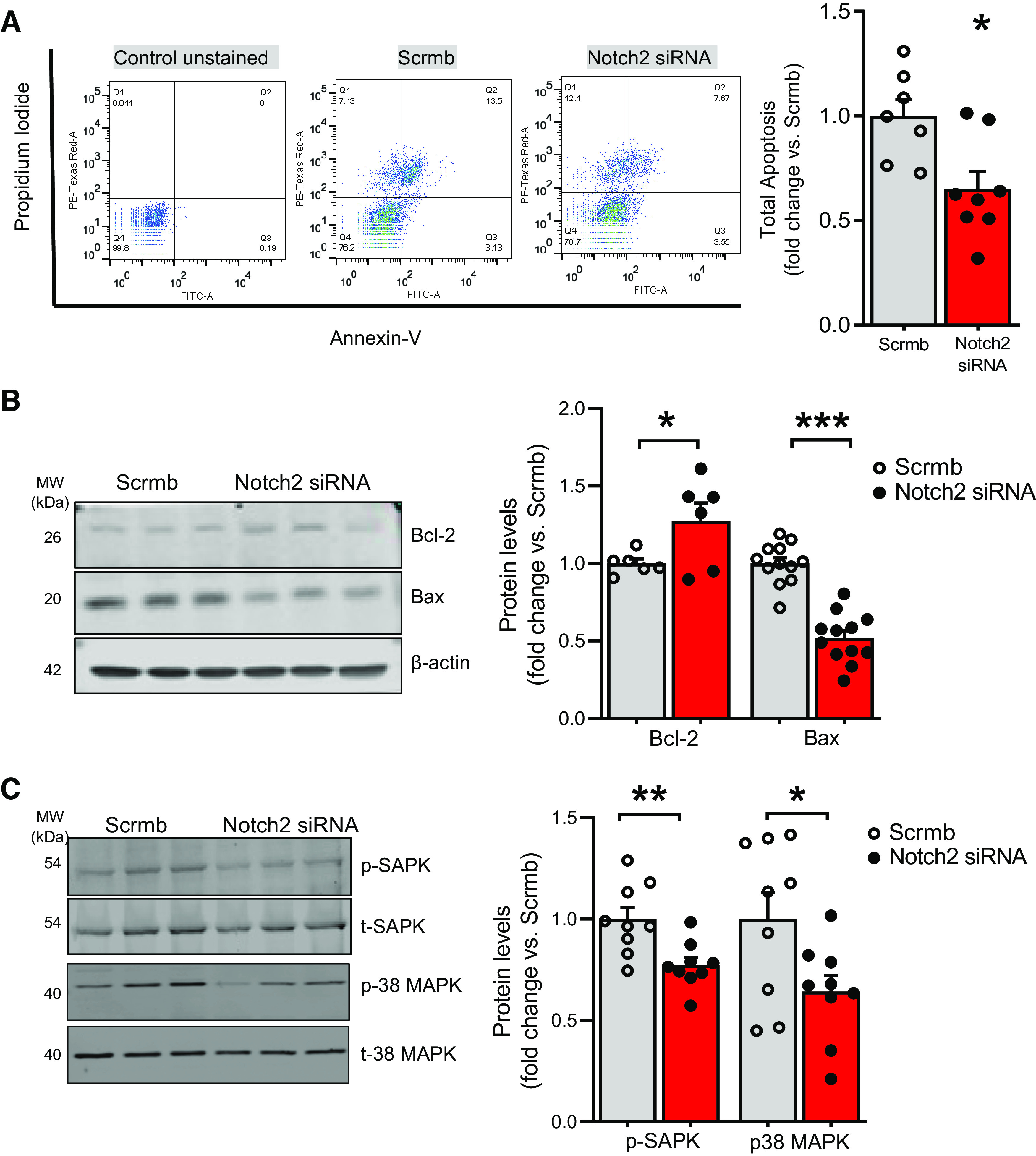
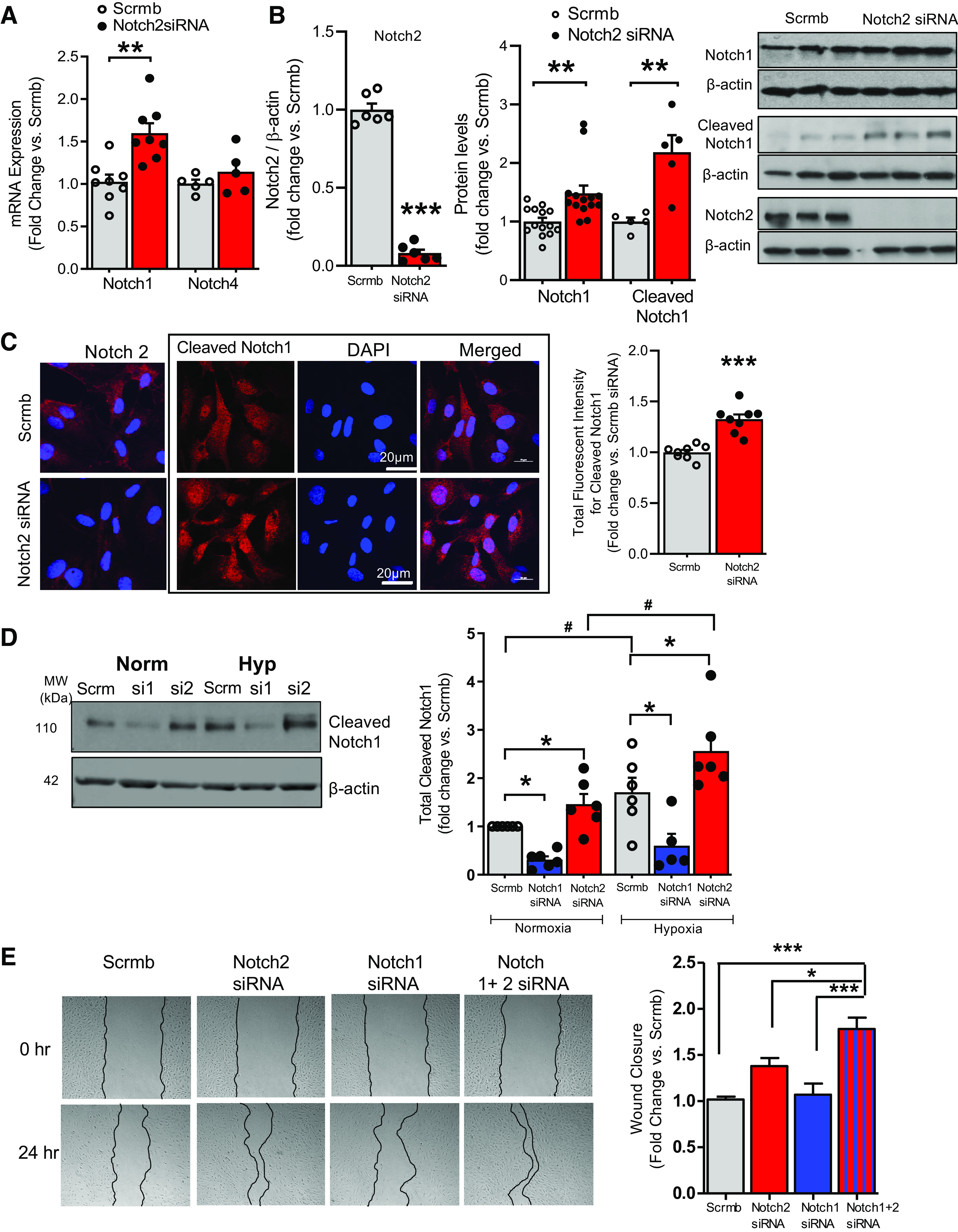
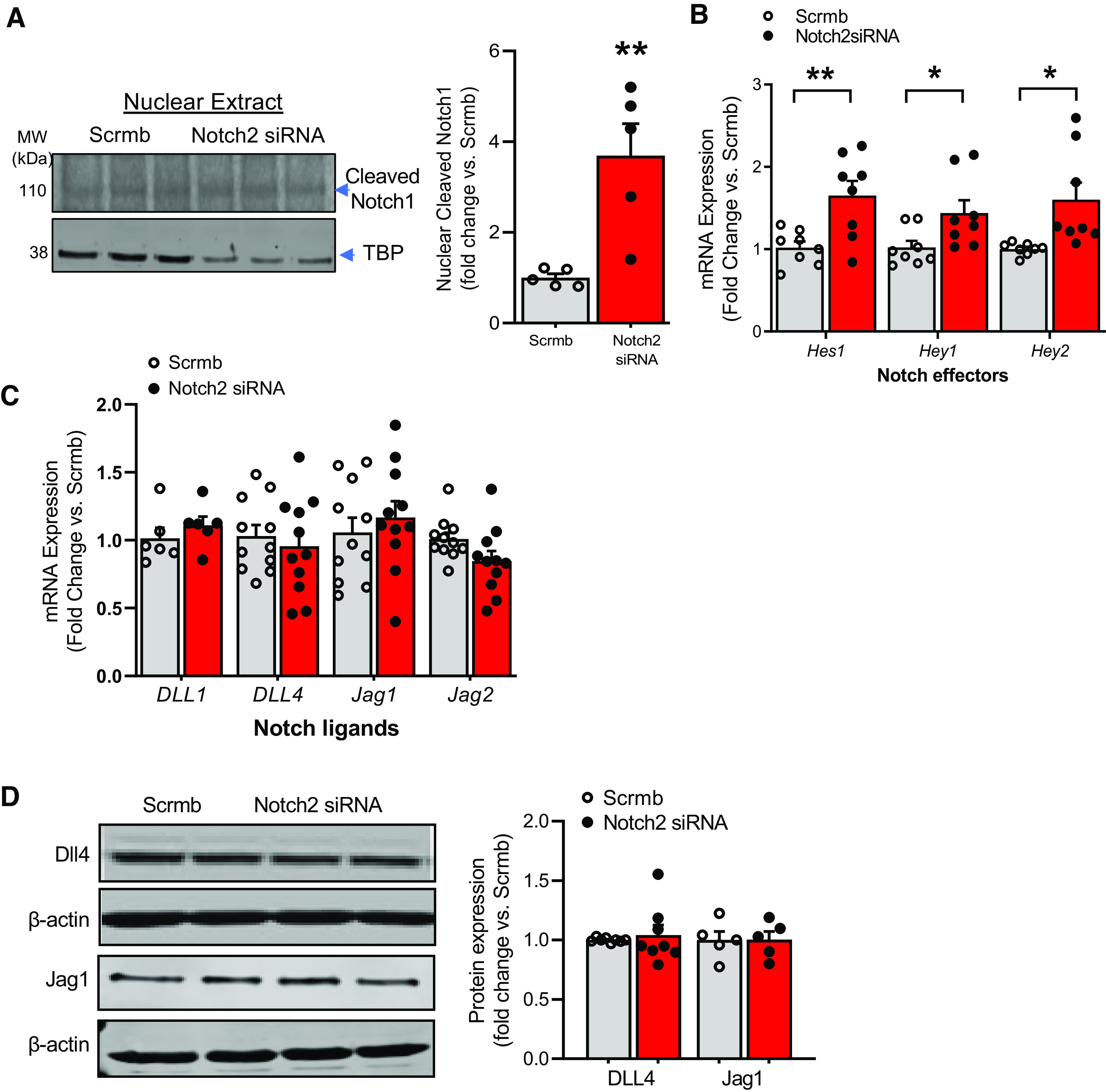
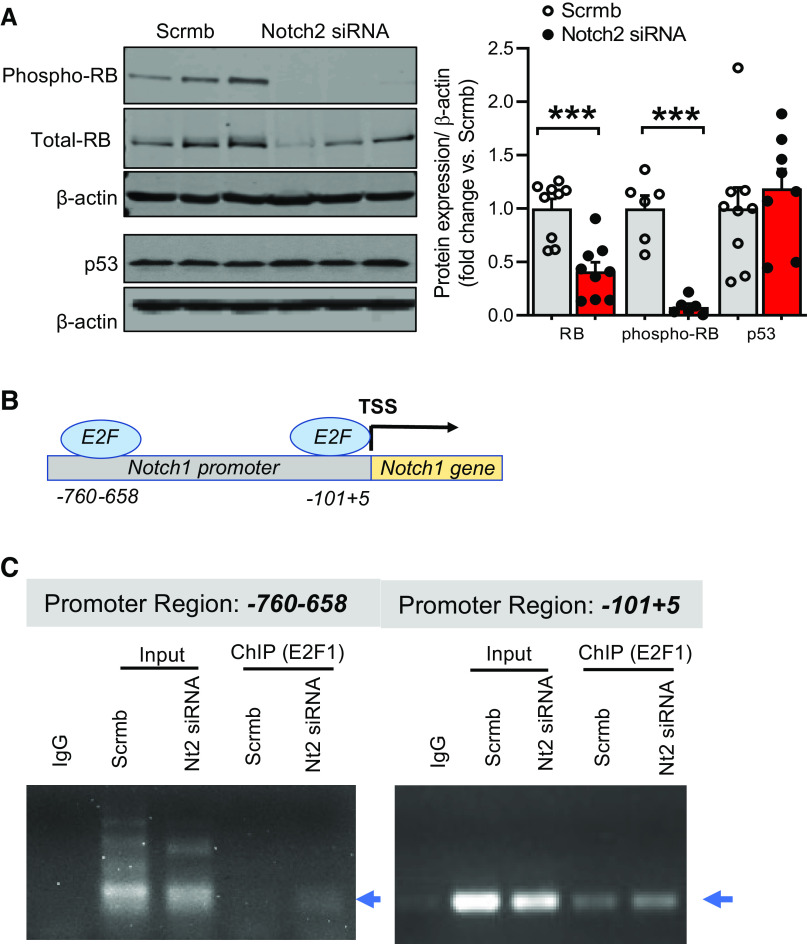
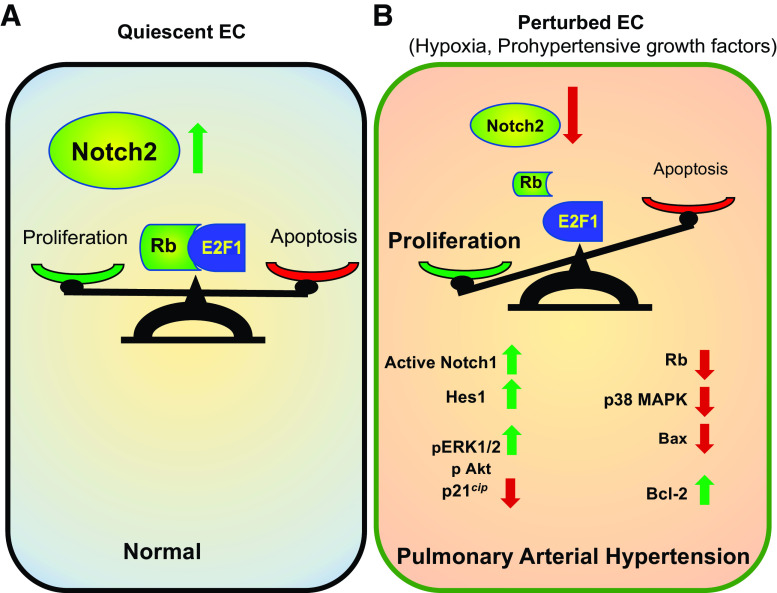
Pulmonary arterial hypertension (PAH) is a fatal cardiopulmonary disease characterized by increased vascular cell proliferation with apoptosis resistance and occlusive remodeling of the small pulmonary arteries. The Notch family of proteins subserves proximal signaling of an evolutionarily conserved pathway that effects cell proliferation, fate determination, and development. In endothelial cells (ECs), Notch receptor 2 (Notch2) was shown to promote endothelial apoptosis. However, a pro- or antiproliferative role for Notch2 in pulmonary endothelial proliferation and ensuing PAH is unknown. We postulated that suppressed Notch2 signaling drives pulmonary endothelial proliferation in the context of PAH. We observed that levels of Notch2 are ablated in lungs from PAH subjects compared with non-PAH controls. Notch2 expression was attenuated in human pulmonary artery endothelial cells (hPAECs) exposed to vasoactive stimuli including hypoxia, TGF-β, ET-1, and IGF-1. Notch2-deficient hPAECs activated Akt, Erk1/2, and antiapoptotic protein Bcl-2 and reduced levels of p21cip and Bax associated with increased EC proliferation and reduced apoptosis. In addition, Notch2 suppression elicited a paradoxical activation of Notch1 and canonical Notch target gene Hes1, Hey1, and Hey2 transcription. Furthermore, reduction in Rb and increased E2F1 binding to the Notch1 promoter appear to explain the Notch1 upregulation. Yet, when Notch1 was decreased in Notch2-suppressed cells, the wound injury response was augmented. In aggregate, our results demonstrate that loss of Notch2 in hPAECs derepresses Notch1 and elicits EC hallmarks of PAH. Augmented EC proliferation upon Notch1 knockdown points to a context-dependent role for Notch1 and 2 in endothelial cell homeostasis.NEW & NOTEWORTHY This study demonstrates a previously unidentified role for Notch2 in the maintenance of lung vascular endothelial cell quiescence and pulmonary artery hypertension (PAH). A key novel finding is that Notch2 suppression activates Notch1 via Rb-E2F1-mediated signaling and induces proliferation and apoptosis resistance in human pulmonary artery endothelial cells. Notably, PAH patients show reduced levels of endothelial Notch2 in their pulmonary arteries, supporting Notch2 as a fundamental driver of PAH pathogenesis.
Keywords: Notch; Rb; endothelial cell; proliferation; pulmonary arterial hypertension.
Figures
Similar articles
-
BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension.Int J Mol Sci. 2024 May 15;25(10):5403. doi: 10.3390/ijms25105403. Int J Mol Sci. 2024. PMID: 38791441 Free PMC article.
-
Inflammatory cytokines induce NOTCH signaling in nucleus pulposus cells: implications in intervertebral disc degeneration.J Biol Chem. 2013 Jun 7;288(23):16761-16774. doi: 10.1074/jbc.M112.446633. Epub 2013 Apr 15. J Biol Chem. 2013. PMID: 23589286 Free PMC article. Clinical Trial.
-
Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension.Eur Respir J. 2016 Oct;48(4):1137-1149. doi: 10.1183/13993003.00773-2015. Epub 2016 Jul 28. Eur Respir J. 2016. PMID: 27471204
-
Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension.Compr Physiol. 2011 Jan;1(1):357-72. doi: 10.1002/cphy.c090005. Compr Physiol. 2011. PMID: 23737177 Free PMC article. Review.
-
Evolving Concepts in Endothelial Pathobiology of Pulmonary Arterial Hypertension.Hypertension. 2022 Aug;79(8):1580-1590. doi: 10.1161/HYPERTENSIONAHA.122.18261. Epub 2022 May 18. Hypertension. 2022. PMID: 35582968 Review.
Cited by
-
Transcription factors and potential therapeutic targets for pulmonary hypertension.Front Cell Dev Biol. 2023 Mar 17;11:1132060. doi: 10.3389/fcell.2023.1132060. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37009479 Free PMC article. Review.
-
Jagged 2 inhibition attenuates hypoxia-induced mitochondrial damage and pulmonary hypertension through Sirtuin 1 signaling.PLoS One. 2024 Jan 26;19(1):e0297525. doi: 10.1371/journal.pone.0297525. eCollection 2024. PLoS One. 2024. PMID: 38277398 Free PMC article.
-
NOTCH3 as a modulator of vascular disease: a target in elastin deficiency and arterial pathologies.J Clin Invest. 2022 Mar 1;132(5):e157007. doi: 10.1172/JCI157007. J Clin Invest. 2022. PMID: 35229725 Free PMC article.
-
Integrated Analysis of Non-Coding RNA and mRNA Expression Profiles in Exosomes from Lung Tissue with Sepsis-Induced Acute Lung Injury.J Inflamm Res. 2023 Sep 1;16:3879-3895. doi: 10.2147/JIR.S419491. eCollection 2023. J Inflamm Res. 2023. PMID: 37674532 Free PMC article.
-
Hemodynamic and Clinical Profiles of Pulmonary Arterial Hypertension Patients with GDF2 and BMPR2 Variants.Int J Mol Sci. 2024 Feb 27;25(5):2734. doi: 10.3390/ijms25052734. Int J Mol Sci. 2024. PMID: 38473983 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL133864/HL/NHLBI NIH HHS/United States
- T32 GM008424/GM/NIGMS NIH HHS/United States
- R01HL142248/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01HL128304/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01HL142638/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL079207/HL/NHLBI NIH HHS/United States
- R01 HL142248/HL/NHLBI NIH HHS/United States
- R01 HL142638/HL/NHLBI NIH HHS/United States
- R01 HL150638/HL/NHLBI NIH HHS/United States
- R01 HL113178/HL/NHLBI NIH HHS/United States
- R01HL112914/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01HL133864/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL112914/HL/NHLBI NIH HHS/United States
- R01HL079207/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL128304/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous