

RNA Helicase DDX17 Inhibits Hepatitis B Virus Replication by Blocking Viral Pregenomic RNA Encapsidation
- PMID: 34287051
- PMCID: PMC8428406
- DOI: 10.1128/JVI.00444-21
RNA Helicase DDX17 Inhibits Hepatitis B Virus Replication by Blocking Viral Pregenomic RNA Encapsidation
Abstract
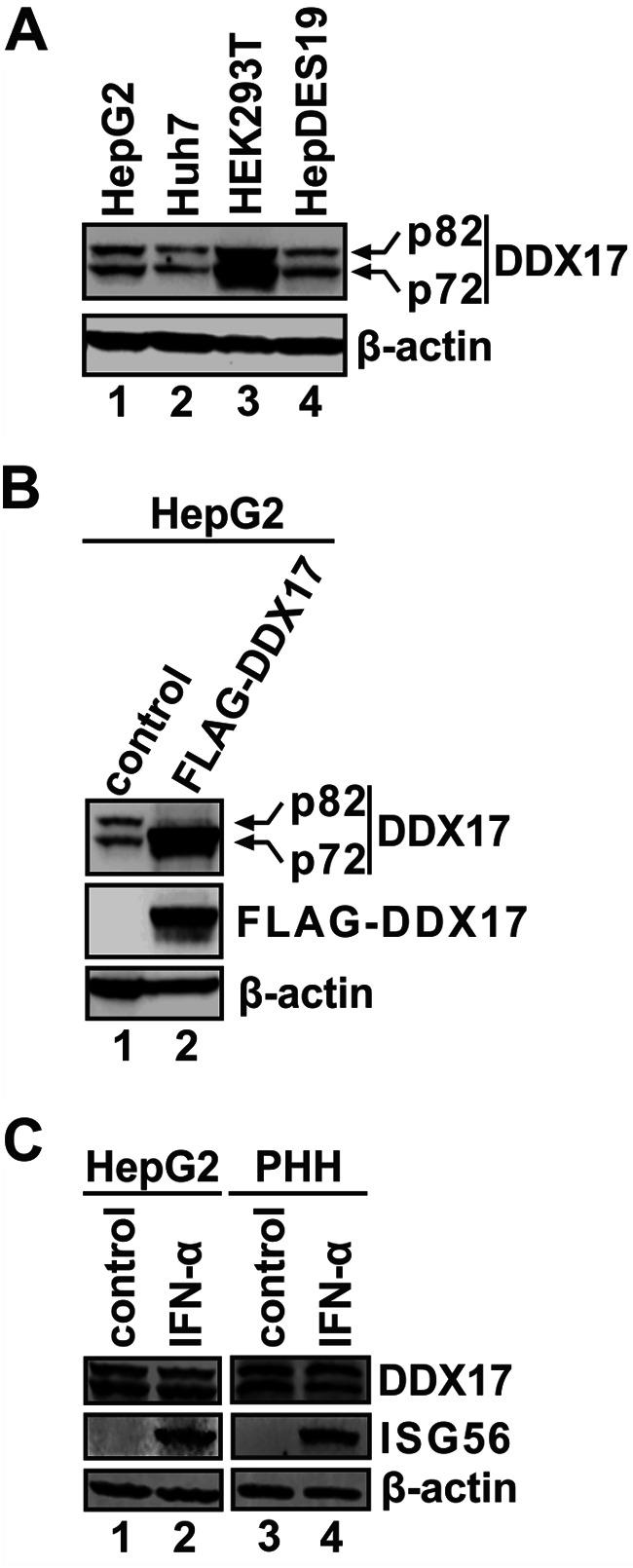
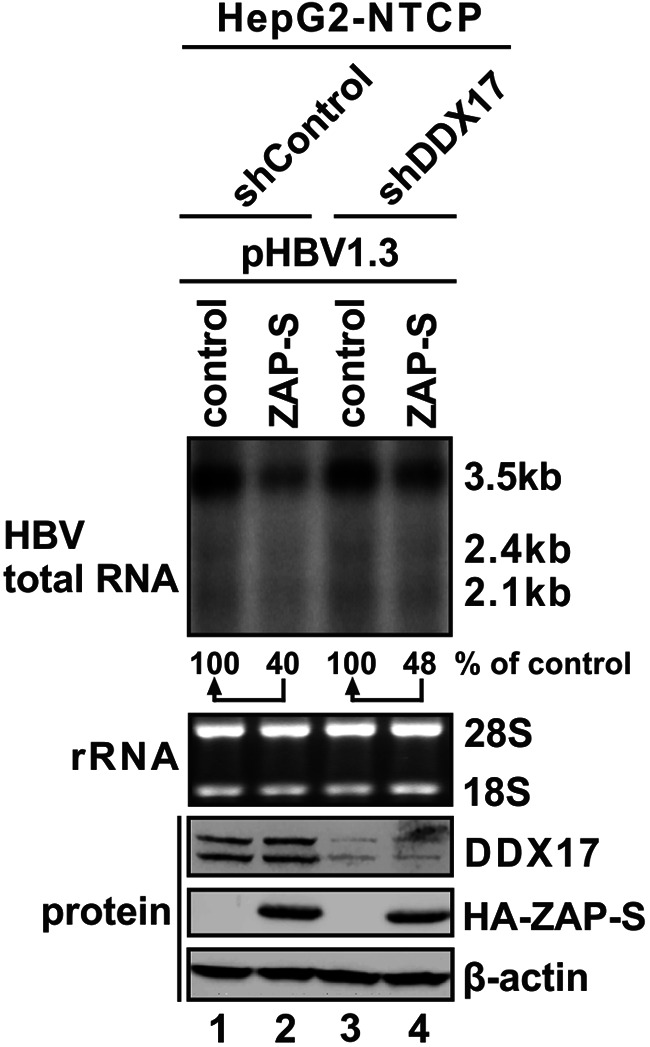
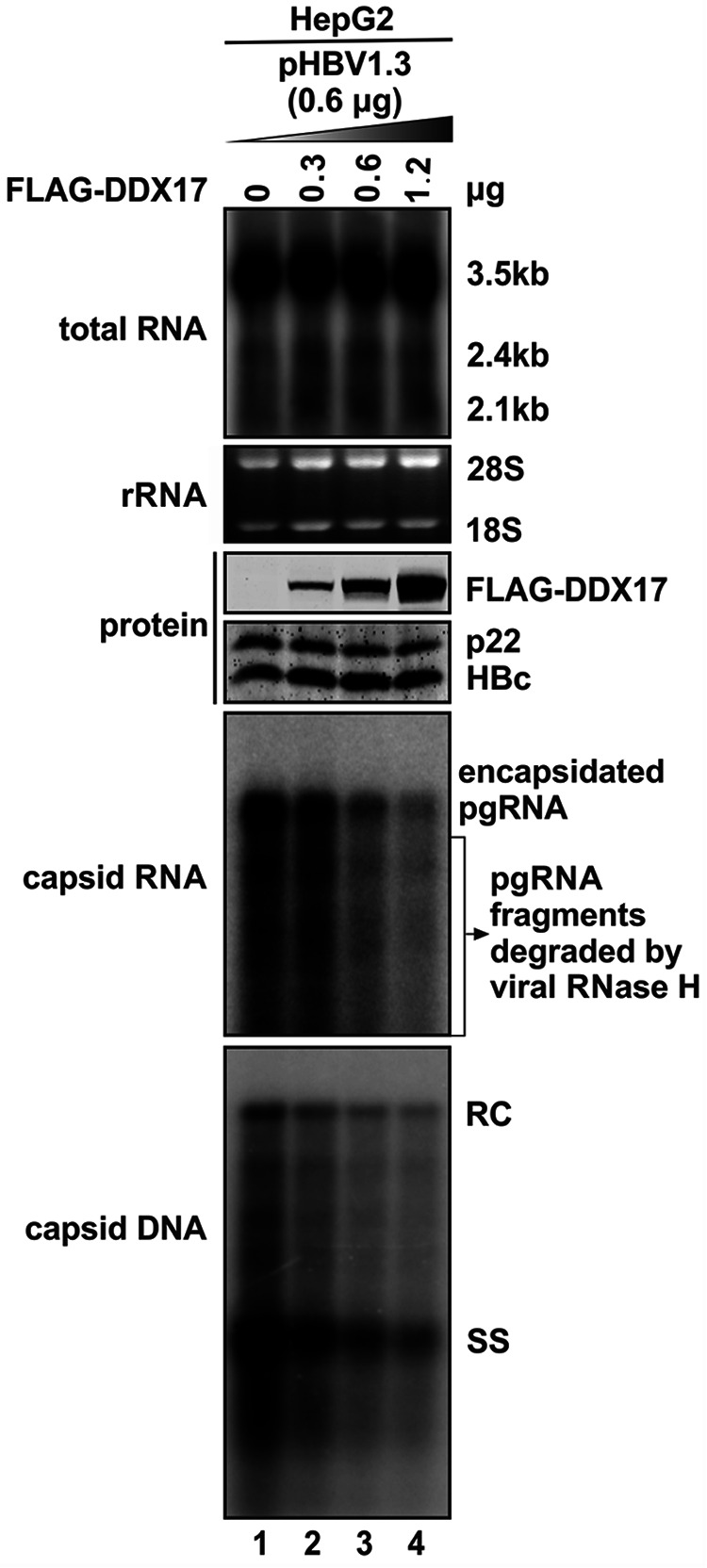
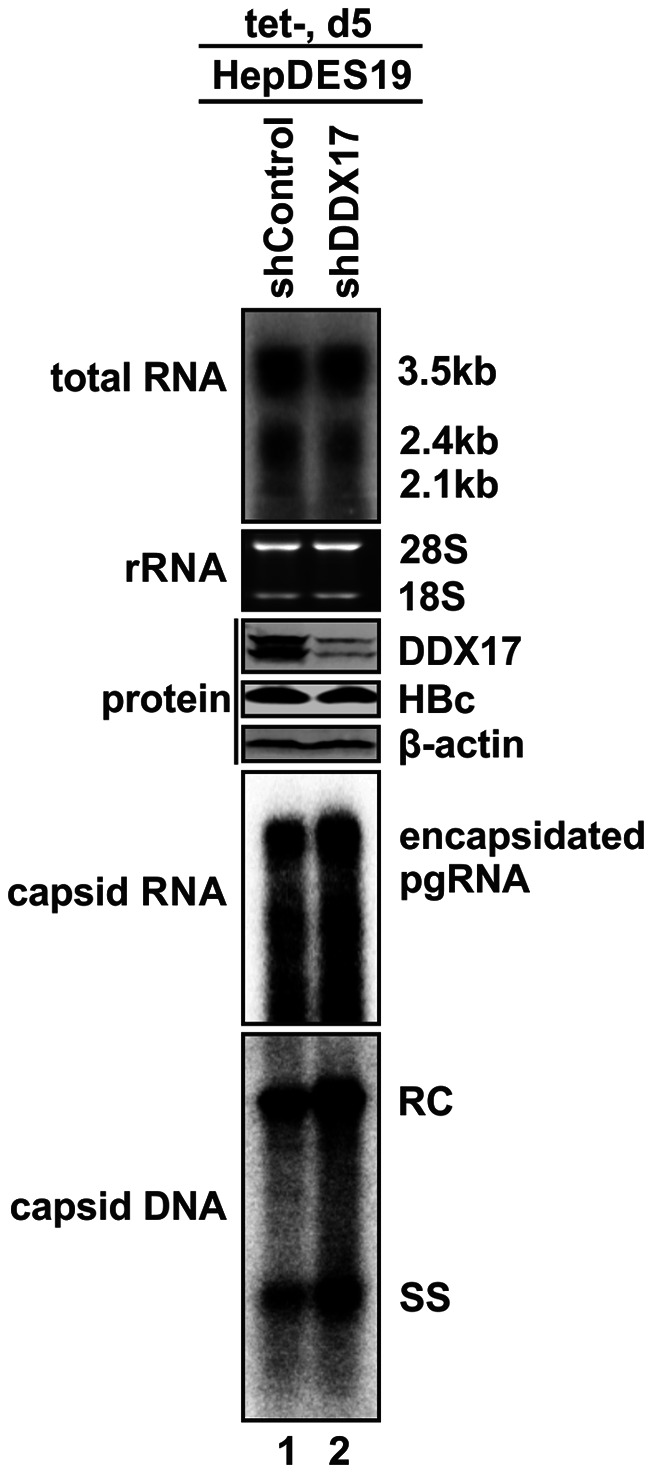
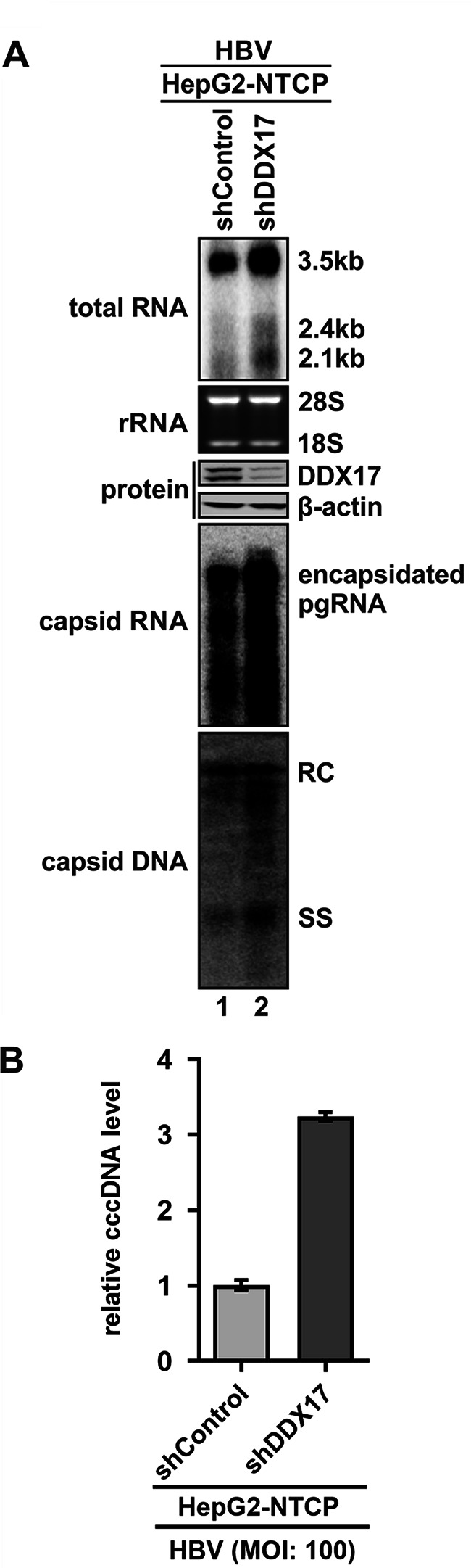
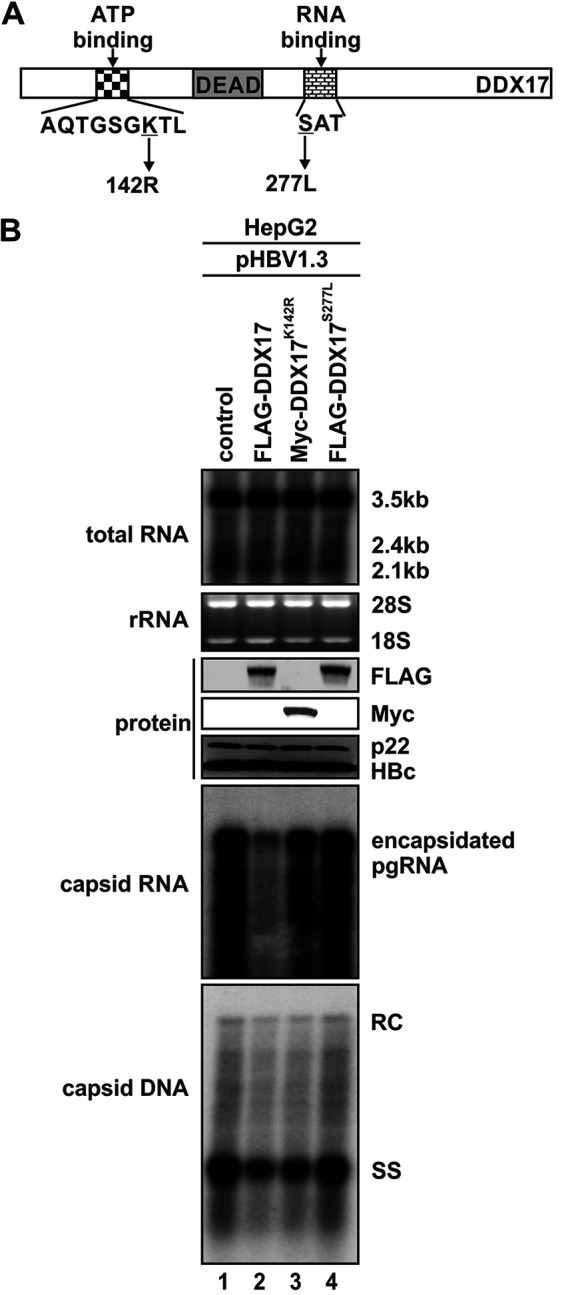
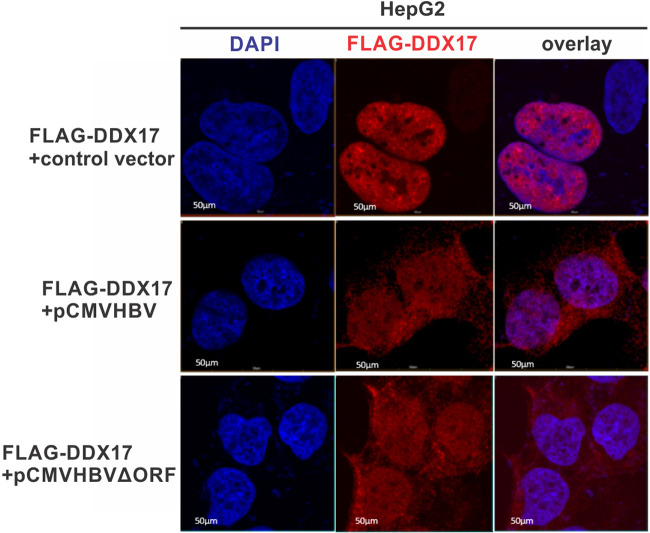
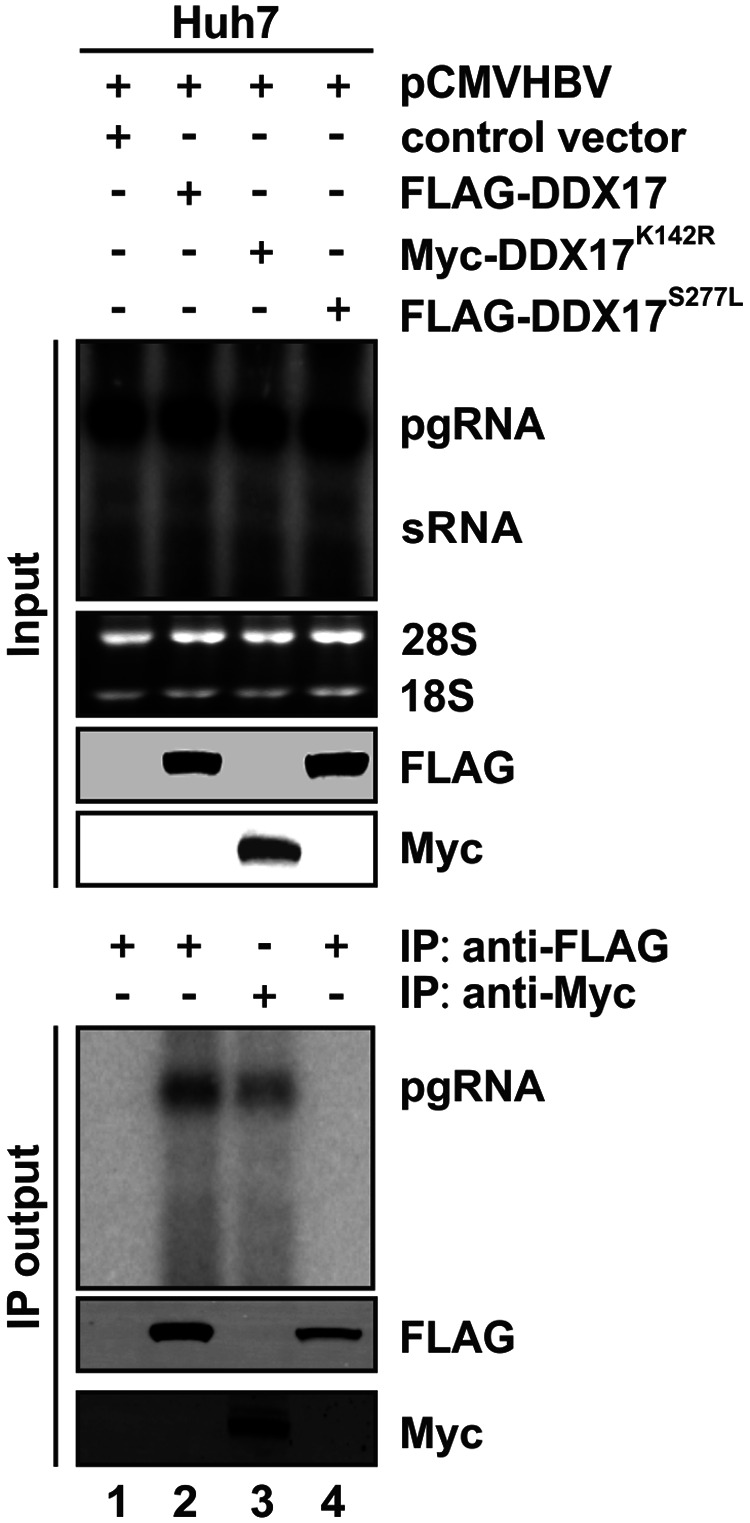
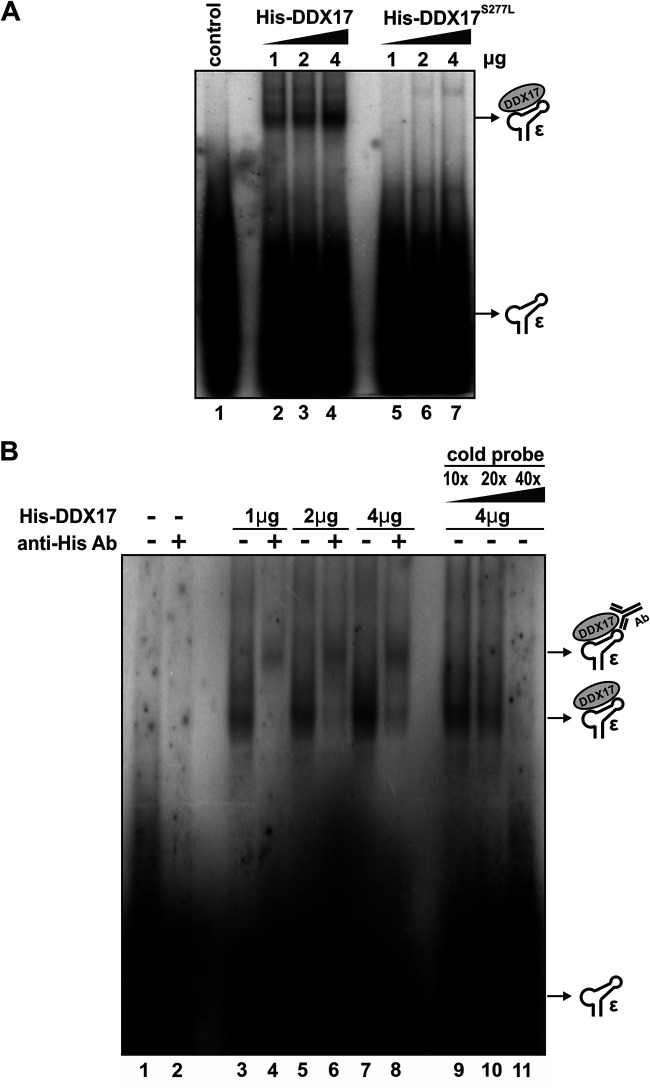
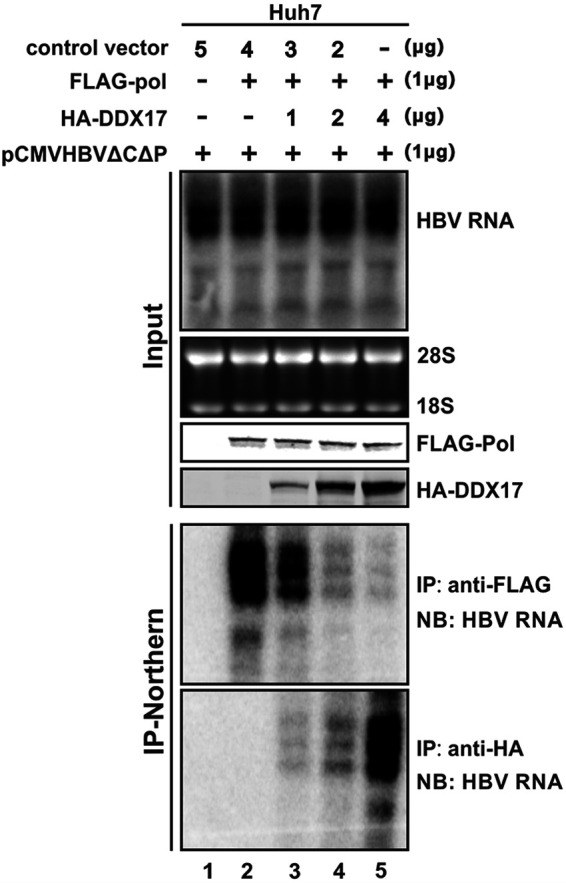
DDX17 is a member of the DEAD-box helicase family proteins involved in cellular RNA folding, splicing, and translation. It has been reported that DDX17 serves as a cofactor of host zinc finger antiviral protein (ZAP)-mediated retroviral RNA degradation and exerts direct antiviral function against Raft Valley fever virus through binding to specific stem-loop structures of viral RNA. Intriguingly, we have previously shown that ZAP inhibits hepatitis B virus (HBV) replication through promoting viral RNA decay, and the ZAP-responsive element (ZRE) of HBV pregenomic RNA (pgRNA) contains a stem-loop structure, specifically epsilon, which serves as the packaging signal for pgRNA encapsidation. In this study, we demonstrated that the endogenous DDX17 is constitutively expressed in human hepatocyte-derived cells but dispensable for ZAP-mediated HBV RNA degradation. However, DDX17 was found to inhibit HBV replication primarily by reducing the level of cytoplasmic encapsidated pgRNA in a helicase-dependent manner. Immunofluorescence assay revealed that DDX17 could gain access to cytoplasm from nucleus in the presence of HBV RNA. In addition, RNA immunoprecipitation and electrophoretic mobility shift assays demonstrated that the enzymatically active DDX17 competes with HBV polymerase to bind to pgRNA at the 5' epsilon motif. In summary, our study suggests that DDX17 serves as an intrinsic host restriction factor against HBV through interfering with pgRNA encapsidation. IMPORTANCE Hepatitis B virus (HBV) chronic infection, a long-studied but yet incurable disease, remains a major public health concern worldwide. Given that HBV replication cycle highly depends on host factors, deepening our understanding of the host-virus interaction is thus of great significance in the journey of finding a cure. In eukaryotic cells, RNA helicases of the DEAD box family are highly conserved enzymes involved in diverse processes of cellular RNA metabolism. Emerging data have shown that DDX17, a typical member of the DEAD box family, functions as an antiviral factor through interacting with viral RNA. In this study, we, for the first time, demonstrate that DDX17 inhibits HBV through blocking the formation of viral replication complex, which not only broadens the antiviral spectrum of DDX17 but also provides new insight into the molecular mechanism of DDX17-mediated virus-host interaction.
Keywords: DDX17; encapsidation; hepatitis B virus; pgRNA; viral replication.
Figures

Similar articles
-
RNA-Binding Motif Protein 24 (RBM24) Is Involved in Pregenomic RNA Packaging by Mediating Interaction between Hepatitis B Virus Polymerase and the Epsilon Element.J Virol. 2019 Mar 5;93(6):e02161-18. doi: 10.1128/JVI.02161-18. Print 2019 Mar 15. J Virol. 2019. PMID: 30626666 Free PMC article.
-
Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein.PLoS Pathog. 2013;9(7):e1003494. doi: 10.1371/journal.ppat.1003494. Epub 2013 Jul 11. PLoS Pathog. 2013. PMID: 23853601 Free PMC article.
-
N6-methyladenosine modification of the 5' epsilon structure of the HBV pregenome RNA regulates its encapsidation by the viral core protein.Proc Natl Acad Sci U S A. 2022 Feb 15;119(7):e2120485119. doi: 10.1073/pnas.2120485119. Proc Natl Acad Sci U S A. 2022. PMID: 35135882 Free PMC article.
-
[Post-transcriptional regulation mechanism and antiviral strategy of hepatitis B virus RNA].Zhonghua Gan Zang Bing Za Zhi. 2024 May 20;32(5):474-480. doi: 10.3760/cma.j.cn501113-20240410-00191. Zhonghua Gan Zang Bing Za Zhi. 2024. PMID: 38858198 Review. Chinese.
-
Pregenomic RNA: How to assist the management of chronic hepatitis B?Rev Med Virol. 2019 Jul;29(4):e2051. doi: 10.1002/rmv.2051. Epub 2019 May 10. Rev Med Virol. 2019. PMID: 31074177 Review.
Cited by
-
Transcriptome and proteomic analysis of mpox virus F3L-expressing cells.Front Cell Infect Microbiol. 2024 Feb 13;14:1354410. doi: 10.3389/fcimb.2024.1354410. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 38415010 Free PMC article.
-
Intracellular Host Restriction of Hepatitis B Virus Replication.Viruses. 2024 May 11;16(5):764. doi: 10.3390/v16050764. Viruses. 2024. PMID: 38793645 Free PMC article. Review.
-
RNA binding protein TIAR modulates HBV replication by tipping the balance of pgRNA translation.Signal Transduct Target Ther. 2023 Sep 13;8(1):346. doi: 10.1038/s41392-023-01573-7. Signal Transduct Target Ther. 2023. PMID: 37699883 Free PMC article.
-
A New Long Noncoding RNA, MAHAT, Inhibits Replication of Porcine Reproductive and Respiratory Syndrome Virus by Recruiting DDX6 To Bind to ZNF34 and Promote an Innate Immune Response.J Virol. 2022 Sep 28;96(18):e0115422. doi: 10.1128/jvi.01154-22. Epub 2022 Sep 8. J Virol. 2022. PMID: 36073922 Free PMC article.
-
Innate Immunity, Inflammation, and Intervention in HBV Infection.Viruses. 2022 Oct 17;14(10):2275. doi: 10.3390/v14102275. Viruses. 2022. PMID: 36298831 Free PMC article. Review.
References
-
- Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, Hu J, Kramvis A, Lampertico P, Janssen HLA, Levrero M, Li W, Liang TJ, Lim SG, Lu F, Penicaud MC, Tavis JE, Thimme R, Zoulim F, Members of the ICE-HBV Working Groups, ICE-HBV Stakeholders Group Chairs, ICE-HBV Senior Advisors. 2019. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol 4:545–558. 10.1016/S2468-1253(19)30119-0. - DOI - PMC - PubMed
-
- Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. 10.7554/eLife.00049. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
