

Macrophages in lung fibrosis
- PMID: 34270737
- PMCID: PMC8633606
- DOI: 10.1093/intimm/dxab040
Macrophages in lung fibrosis
Abstract
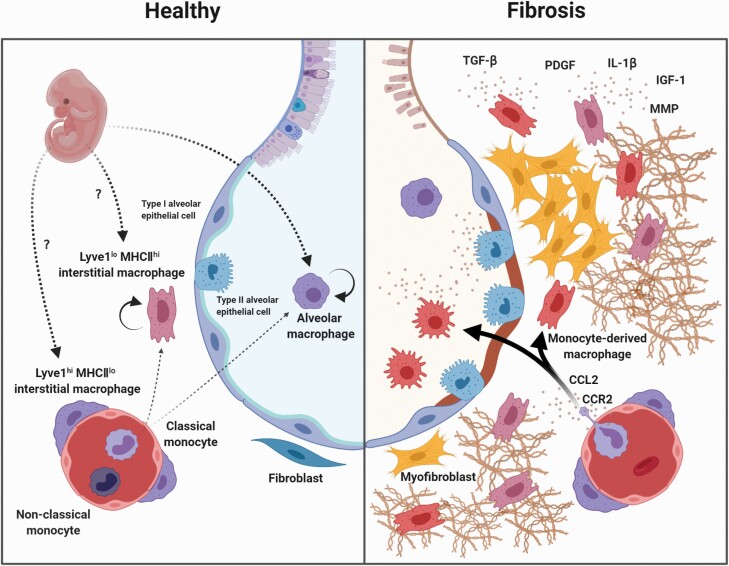
Pulmonary fibrosis (PF) is a disease in which excessive extracellular matrix (ECM) accumulation occurs in the lungs, which induces thickening of the alveolar walls, ultimately leading to the destruction of alveolar structures and respiratory failure. Idiopathic PF, the cause of which is unknown, has a poor prognosis with a median survival of 2-4 years after diagnosis. There is currently no known curative treatment. The mechanism underlying PF is thought to be initiated by the dysfunction of type II alveolar epithelial cells, which leads to ECM overproduction through the activation of fibroblasts. In addition, it has been suggested that a variety of cells contribute to fibrotic processes. In particular, clinical and basic research findings examining the roles of macrophages suggest that they may be pivotal regulators of PF. In this review, we discuss the characteristics, functions and origins of subsets of macrophages involved in PF, including resident alveolar, interstitial and monocyte-derived macrophages.
Keywords: chemokine; fibroblasts; growth factor; interstitial lung disease; single-cell transcriptome.
© The Author(s) 2021. Published by Oxford University Press on behalf of The Japanese Society for Immunology.
Figures
Similar articles
-
Human pluripotent stem cell-derived macrophages and macrophage-derived exosomes: therapeutic potential in pulmonary fibrosis.Stem Cell Res Ther. 2022 Sep 2;13(1):433. doi: 10.1186/s13287-022-03136-z. Stem Cell Res Ther. 2022. PMID: 36056418 Free PMC article. Review.
-
Macrophages in Lung Repair and Fibrosis.Results Probl Cell Differ. 2024;74:257-290. doi: 10.1007/978-3-031-65944-7_10. Results Probl Cell Differ. 2024. PMID: 39406909 Review.
-
IRAK-M Regulates Monocyte Trafficking to the Lungs in Response to Bleomycin Challenge.J Immunol. 2020 May 15;204(10):2661-2670. doi: 10.4049/jimmunol.1900466. Epub 2020 Apr 6. J Immunol. 2020. PMID: 32253243 Free PMC article.
-
Regulation of alveolar macrophage death in pulmonary fibrosis: a review.Apoptosis. 2023 Dec;28(11-12):1505-1519. doi: 10.1007/s10495-023-01888-4. Epub 2023 Sep 14. Apoptosis. 2023. PMID: 37707713 Free PMC article. Review.
-
S100a4 Is Secreted by Alternatively Activated Alveolar Macrophages and Promotes Activation of Lung Fibroblasts in Pulmonary Fibrosis.Front Immunol. 2018 Jun 1;9:1216. doi: 10.3389/fimmu.2018.01216. eCollection 2018. Front Immunol. 2018. PMID: 29910813 Free PMC article.
Cited by
-
Direct M2 macrophage co-culture overrides viscoelastic hydrogel mechanics to promote fibroblast activation.bioRxiv [Preprint]. 2024 Oct 15:2024.10.13.618034. doi: 10.1101/2024.10.13.618034. bioRxiv. 2024. PMID: 39463963 Free PMC article. Preprint.
-
Toxicogenomic assessment of in vitro macrophages exposed to profibrotic challenge reveals a sustained transcriptomic immune signature.Comput Struct Biotechnol J. 2024 Oct 8;25:194-204. doi: 10.1016/j.csbj.2024.10.010. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 39430886 Free PMC article.
-
Reproducible lung protective effects of a TGFβR1/ALK5 inhibitor in a bleomycin-induced and spirometry-confirmed model of IPF in male mice.Physiol Rep. 2024 Oct;12(19):e70077. doi: 10.14814/phy2.70077. Physiol Rep. 2024. PMID: 39394052 Free PMC article.
-
Transformation of macrophages into myofibroblasts in fibrosis-related diseases: emerging biological concepts and potential mechanism.Front Immunol. 2024 Sep 25;15:1474688. doi: 10.3389/fimmu.2024.1474688. eCollection 2024. Front Immunol. 2024. PMID: 39386212 Free PMC article. Review.
-
BRD4: an effective target for organ fibrosis.Biomark Res. 2024 Aug 30;12(1):92. doi: 10.1186/s40364-024-00641-6. Biomark Res. 2024. PMID: 39215370 Free PMC article. Review.
