

Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy
- PMID: 34208776
- PMCID: PMC8234207
- DOI: 10.3390/cells10061521
Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy
Abstract
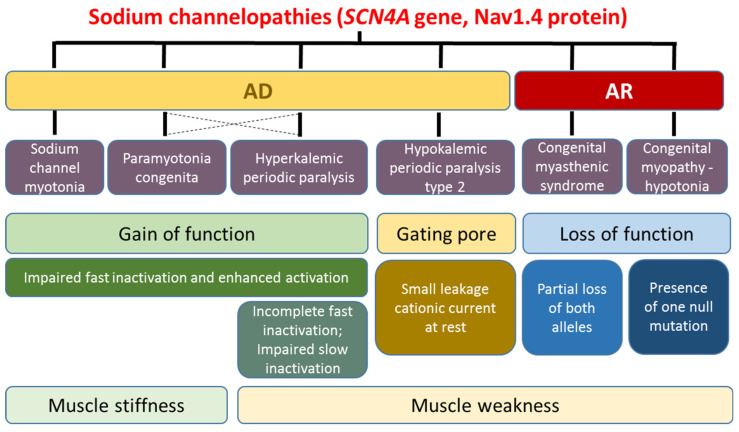
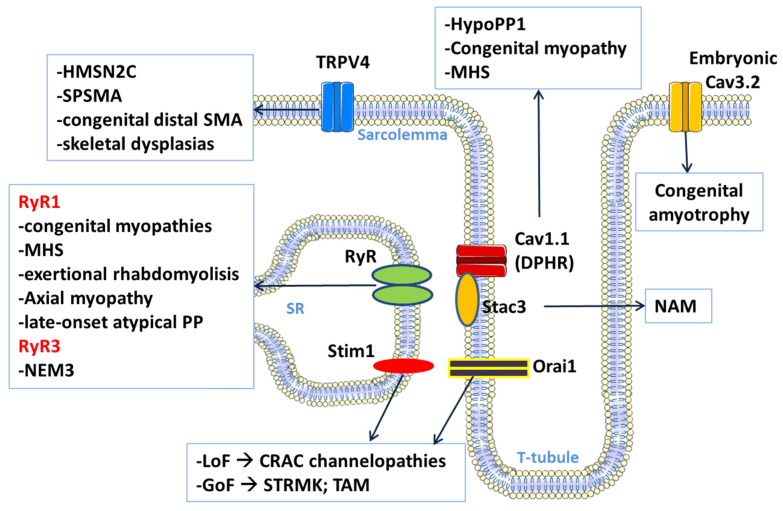
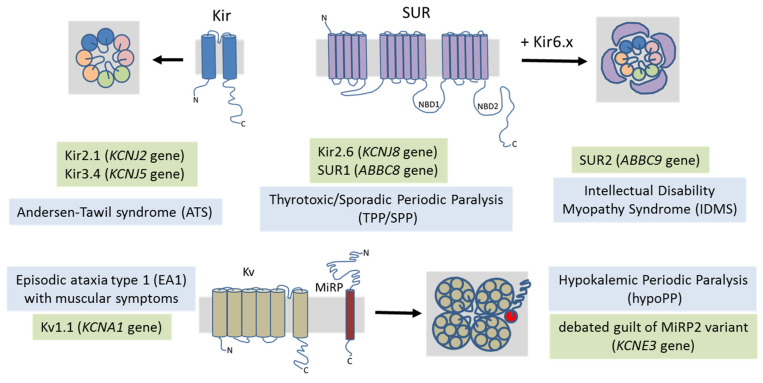
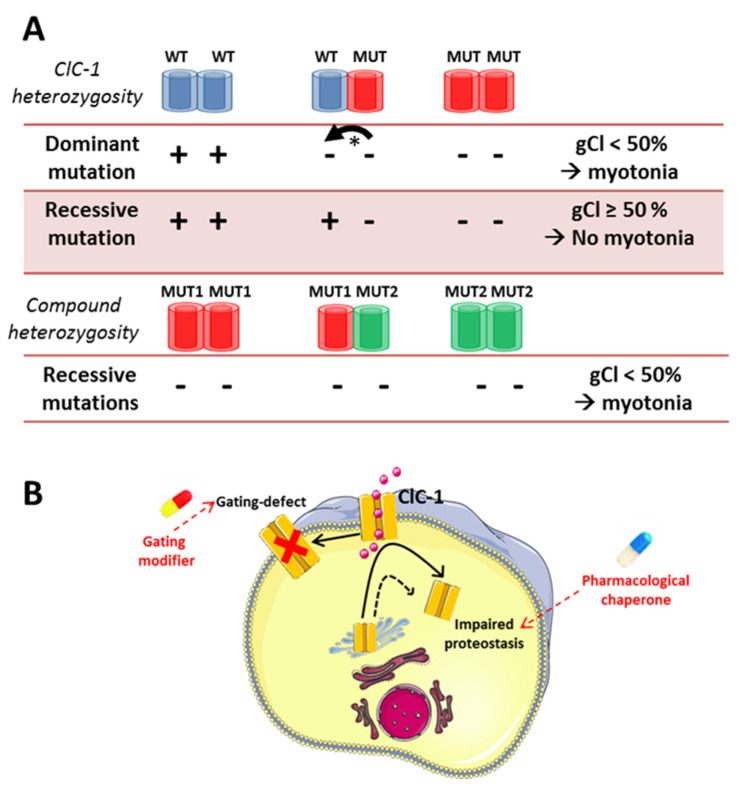
Skeletal muscle ion channelopathies (SMICs) are a large heterogeneous group of rare genetic disorders caused by mutations in genes encoding ion channel subunits in the skeletal muscle mainly characterized by myotonia or periodic paralysis, potentially resulting in long-term disabilities. However, with the development of new molecular technologies, new genes and new phenotypes, including progressive myopathies, have been recently discovered, markedly increasing the complexity in the field. In this regard, new advances in SMICs show a less conventional role of ion channels in muscle cell division, proliferation, differentiation, and survival. Hence, SMICs represent an expanding and exciting field. Here, we review current knowledge of SMICs, with a description of their clinical phenotypes, cellular and molecular pathomechanisms, and available treatments.
Keywords: CACNA1S; CLCN1; KCNJ2; SCN4A; ion channels; myopathies; myotonia; periodic paralysis.
Conflict of interest statement
The authors report no disclosure relevant to the manuscript.
Figures
Similar articles
-
Mutation spectrum and health status in skeletal muscle channelopathies in Japan.Neuromuscul Disord. 2020 Jul;30(7):546-553. doi: 10.1016/j.nmd.2020.06.001. Epub 2020 Jun 7. Neuromuscul Disord. 2020. PMID: 32660787
-
Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands.Neuromuscul Disord. 2018 May;28(5):402-407. doi: 10.1016/j.nmd.2018.03.006. Epub 2018 Mar 9. Neuromuscul Disord. 2018. PMID: 29606556
-
Novel insights into the pathomechanisms of skeletal muscle channelopathies.Curr Neurol Neurosci Rep. 2012 Feb;12(1):62-9. doi: 10.1007/s11910-011-0238-3. Curr Neurol Neurosci Rep. 2012. PMID: 22083238 Review.
-
Prevalence study of genetically defined skeletal muscle channelopathies in England.Neurology. 2013 Apr 16;80(16):1472-5. doi: 10.1212/WNL.0b013e31828cf8d0. Epub 2013 Mar 20. Neurology. 2013. PMID: 23516313 Free PMC article.
-
Pathomechanisms in channelopathies of skeletal muscle and brain.Annu Rev Neurosci. 2006;29:387-415. doi: 10.1146/annurev.neuro.29.051605.112815. Annu Rev Neurosci. 2006. PMID: 16776591 Review.
Cited by
-
Blockers of Skeletal Muscle Nav1.4 Channels: From Therapy of Myotonic Syndrome to Molecular Determinants of Pharmacological Action and Back.Int J Mol Sci. 2023 Jan 3;24(1):857. doi: 10.3390/ijms24010857. Int J Mol Sci. 2023. PMID: 36614292 Free PMC article. Review.
-
Unveiling the neuroprotective potential of dietary polysaccharides: a systematic review.Front Nutr. 2023 Nov 22;10:1299117. doi: 10.3389/fnut.2023.1299117. eCollection 2023. Front Nutr. 2023. PMID: 38075226 Free PMC article. Review.
-
A c.1775C > T Point Mutation of Sodium Channel Alfa Subunit Gene (SCN4A) in a Three-Generation Sardinian Family with Sodium Channel Myotonia.J Neuromuscul Dis. 2024;11(3):725-734. doi: 10.3233/JND-230134. J Neuromuscul Dis. 2024. PMID: 38427496 Free PMC article.
-
Chaperone activity of niflumic acid on ClC-1 chloride channel mutants causing myotonia congenita.Front Pharmacol. 2022 Aug 11;13:958196. doi: 10.3389/fphar.2022.958196. eCollection 2022. Front Pharmacol. 2022. PMID: 36034862 Free PMC article.
-
Genetic analysis of 37 cases with primary periodic paralysis in Chinese patients.Orphanet J Rare Dis. 2024 Apr 12;19(1):160. doi: 10.1186/s13023-024-03170-5. Orphanet J Rare Dis. 2024. PMID: 38609989 Free PMC article.
References
-
- Stunnenberg B.C., Raaphorst J., Deenen J.C., Links T.P., Wilde A.A., Verbove D.J., Kamsteeg E.J., van den Wijngaard A., Faber C.G., van der Wilt G.J., et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul. Disord. 2018;28:402–407. doi: 10.1016/j.nmd.2018.03.006. - DOI - PubMed
-
- Maggi L., Brugnoni R., Canioni E., Tonin P., Saletti V., Patrizia S., Cotti Piccinelli S., Colleoni L., Ferrigno P., Antonella P., et al. Clinical and Molecular Spectrum of Myotonia and Periodic Paralyses Associated With Mutations in SCN4A in a Large Cohort of Italian Patients. Front. Neurol. 2020;11:646. doi: 10.3389/fneur.2020.00646. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
