

Mind the replication gap
- PMID: 34113447
- PMCID: PMC8188003
- DOI: 10.1098/rsos.201932
Mind the replication gap
Abstract
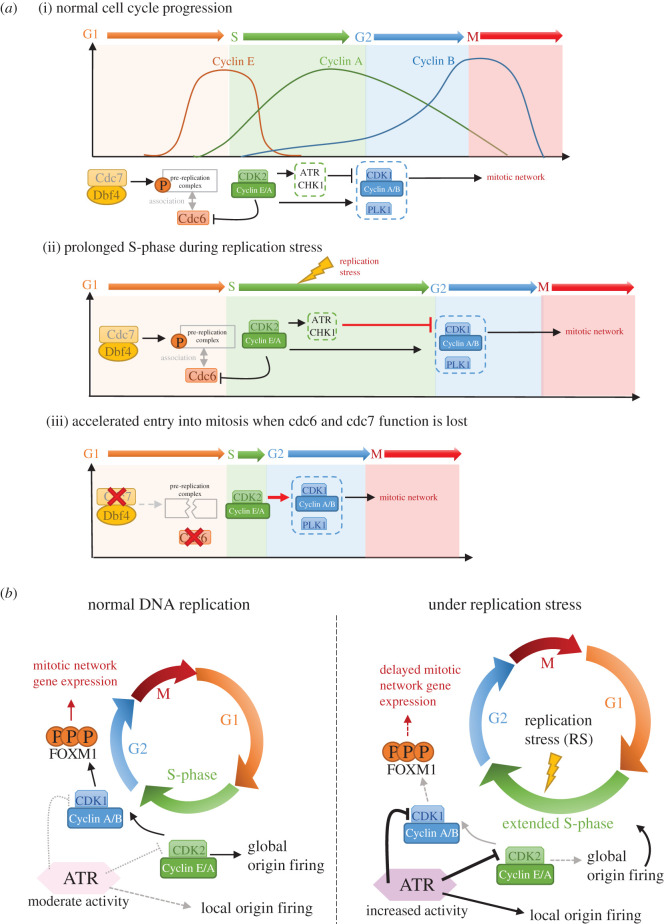
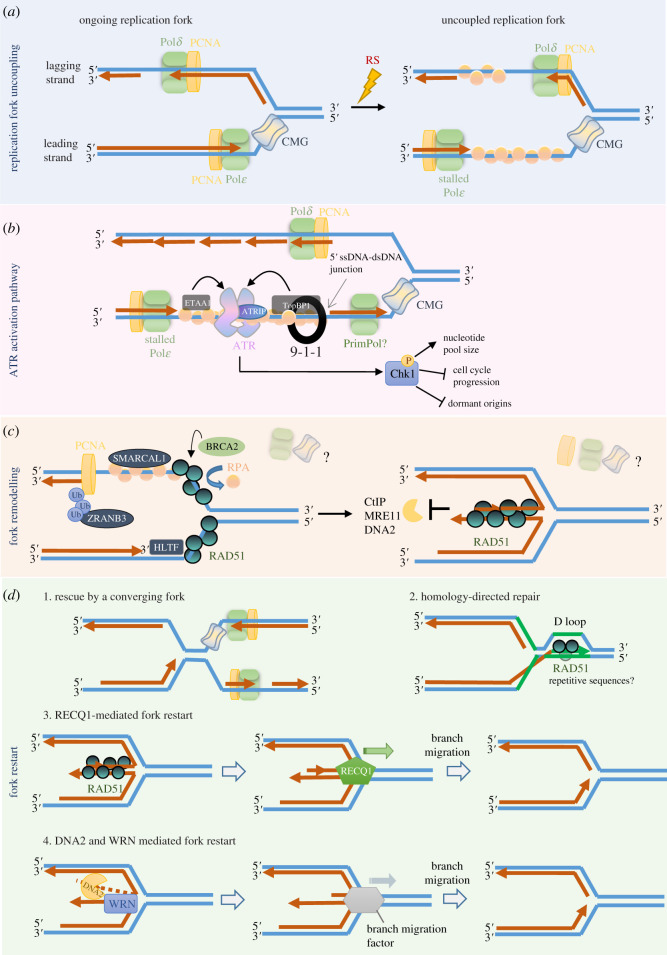
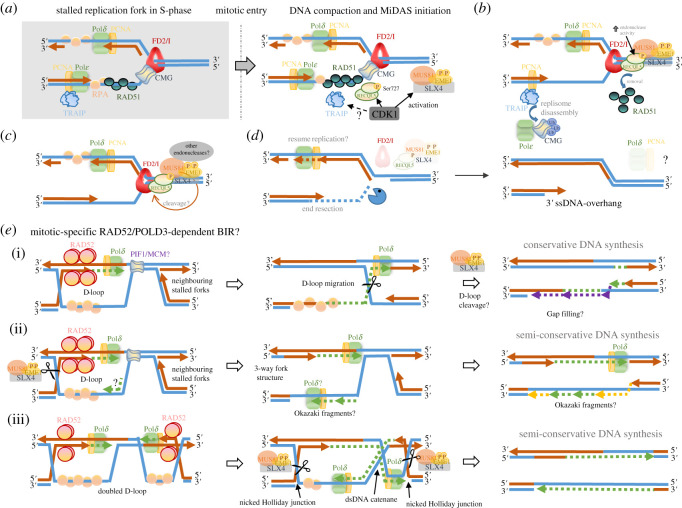
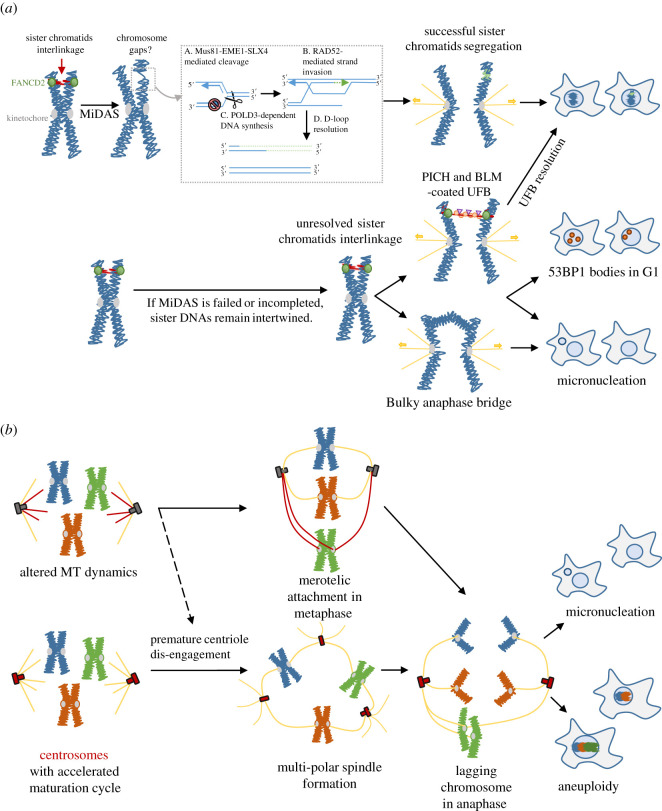
Unlike bacteria, mammalian cells need to complete DNA replication before segregating their chromosomes for the maintenance of genome integrity. Thus, cells have evolved efficient pathways to restore stalled and/or collapsed replication forks during S-phase, and when necessary, also to delay cell cycle progression to ensure replication completion. However, strong evidence shows that cells can proceed to mitosis with incompletely replicated DNA when under mild replication stress (RS) conditions. Consequently, the incompletely replicated genomic gaps form, predominantly at common fragile site regions, where the converging fork-like DNA structures accumulate. These branched structures pose a severe threat to the faithful disjunction of chromosomes as they physically interlink the partially duplicated sister chromatids. In this review, we provide an overview discussing how cells respond and deal with the under-replicated DNA structures that escape from the S/G2 surveillance system. We also focus on recent research of a mitotic break-induced replication pathway (also known as mitotic DNA repair synthesis), which has been proposed to operate during prophase in an attempt to finish DNA synthesis at the under-replicated genomic regions. Finally, we discuss recent data on how mild RS may cause chromosome instability and mutations that accelerate cancer genome evolution.
Keywords: break-induced replication; chromosome instability; common fragile sites; mitotic DNA repair synthesis; replication stress; ultrafine DNA bridges.
© 2021 The Authors.
Figures
Similar articles
-
When RAD52 Allows Mitosis to Accept Unscheduled DNA Synthesis.Cancers (Basel). 2019 Dec 19;12(1):26. doi: 10.3390/cancers12010026. Cancers (Basel). 2019. PMID: 31861741 Free PMC article. Review.
-
Under-Replicated DNA: The Byproduct of Large Genomes?Cancers (Basel). 2020 Sep 25;12(10):2764. doi: 10.3390/cancers12102764. Cancers (Basel). 2020. PMID: 32992928 Free PMC article. Review.
-
Inducing and Detecting Mitotic DNA Synthesis at Difficult-to-Replicate Loci.Methods Enzymol. 2018;601:45-58. doi: 10.1016/bs.mie.2017.11.025. Epub 2018 Feb 3. Methods Enzymol. 2018. PMID: 29523241 Review.
-
Cohesin dynamic association to chromatin and interfacing with replication forks in genome integrity maintenance.Curr Genet. 2018 Oct;64(5):1005-1013. doi: 10.1007/s00294-018-0824-x. Epub 2018 Mar 16. Curr Genet. 2018. PMID: 29549581 Review.
-
Replication Stress Response Links RAD52 to Protecting Common Fragile Sites.Cancers (Basel). 2019 Sep 29;11(10):1467. doi: 10.3390/cancers11101467. Cancers (Basel). 2019. PMID: 31569559 Free PMC article. Review.
Cited by
-
Role of Cockayne Syndrome Group B Protein in Replication Stress: Implications for Cancer Therapy.Int J Mol Sci. 2022 Sep 6;23(18):10212. doi: 10.3390/ijms231810212. Int J Mol Sci. 2022. PMID: 36142121 Free PMC article. Review.
-
Mistimed origin licensing and activation stabilize common fragile sites under tight DNA-replication checkpoint activation.Nat Struct Mol Biol. 2023 Apr;30(4):539-550. doi: 10.1038/s41594-023-00949-1. Epub 2023 Apr 6. Nat Struct Mol Biol. 2023. PMID: 37024657
-
CSB and SMARCAL1 compete for RPA32 at stalled forks and differentially control the fate of stalled forks in BRCA2-deficient cells.Nucleic Acids Res. 2024 May 22;52(9):5067-5087. doi: 10.1093/nar/gkae154. Nucleic Acids Res. 2024. PMID: 38416570 Free PMC article.
-
Life Sciences New Talent collection.R Soc Open Sci. 2022 Jan 19;9(1):211981. doi: 10.1098/rsos.211981. eCollection 2022 Jan. R Soc Open Sci. 2022. PMID: 35116170 Free PMC article. No abstract available.
-
Microscopic Detection of DNA Synthesis in Early Mitosis at Repetitive lacO Sequences in Human Cells.Bio Protoc. 2022 Sep 5;12(17):e4504. doi: 10.21769/BioProtoc.4504. eCollection 2022 Sep 5. Bio Protoc. 2022. PMID: 36213106 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
