

Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein
- PMID: 34084427
- PMCID: PMC8115088
- DOI: 10.1039/d0sc06288j
Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein
Abstract
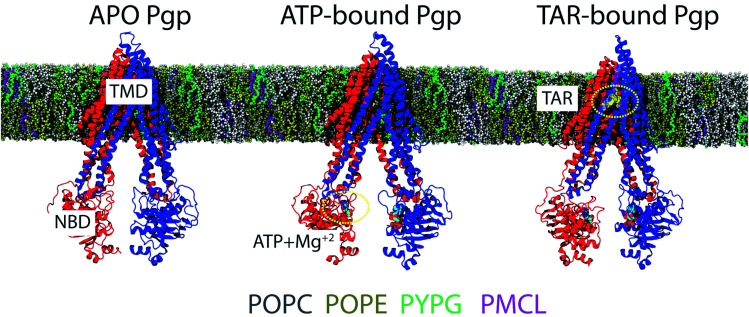
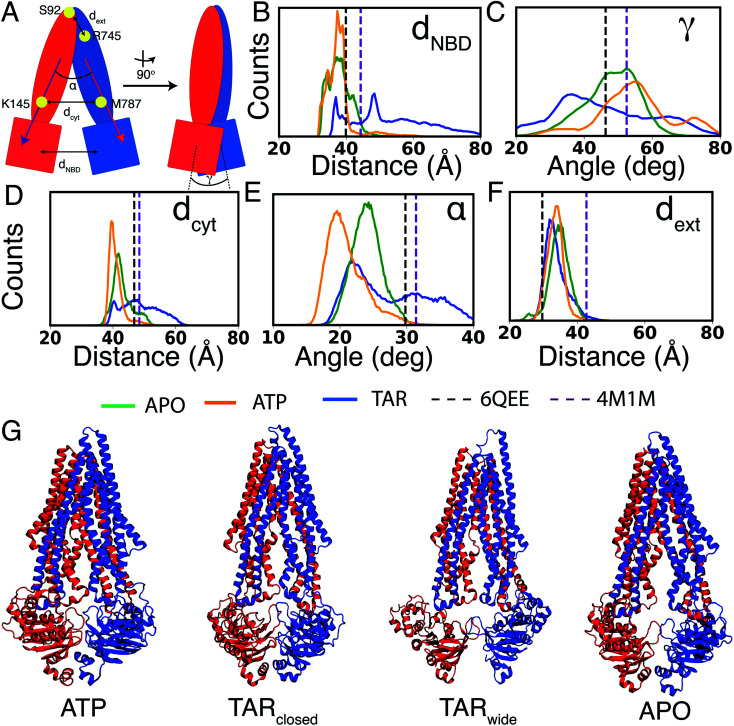
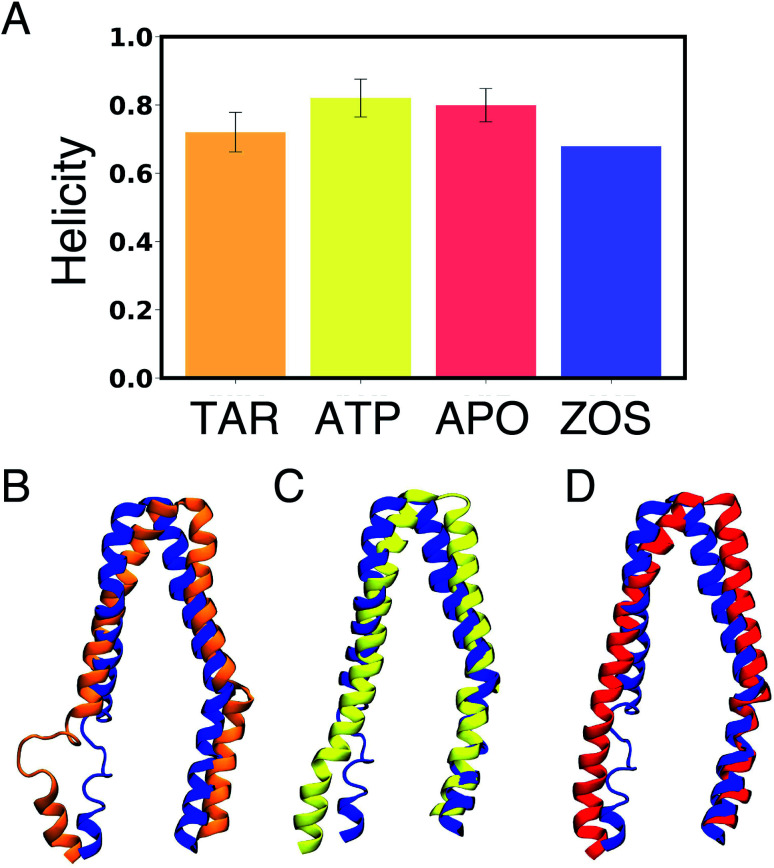
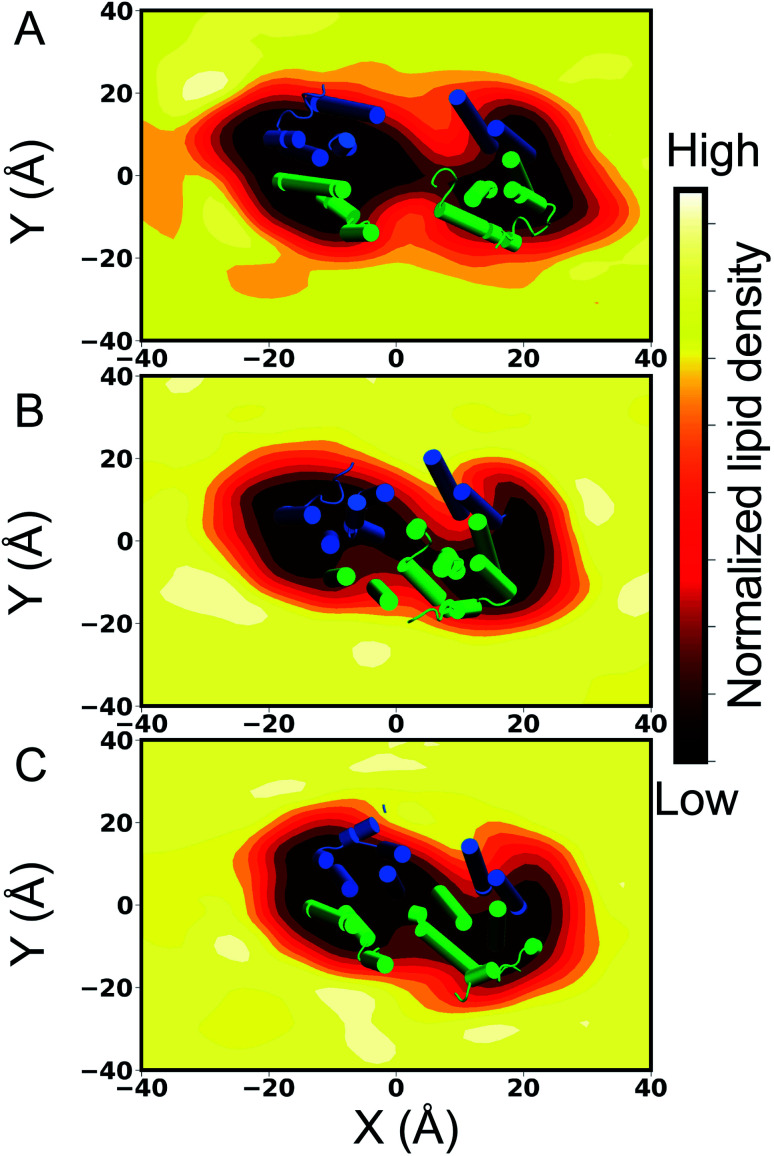
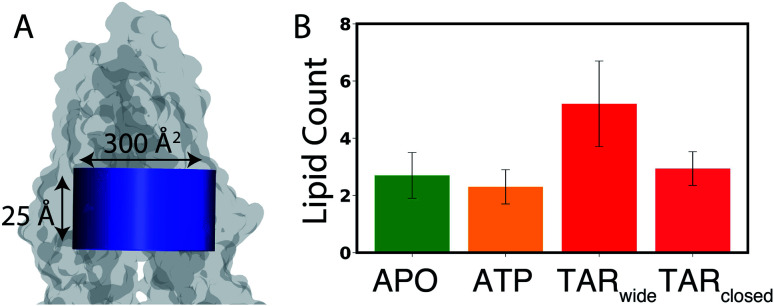
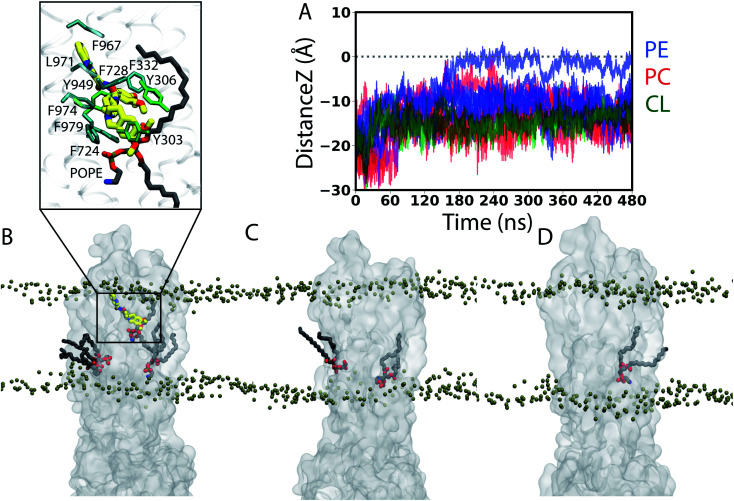
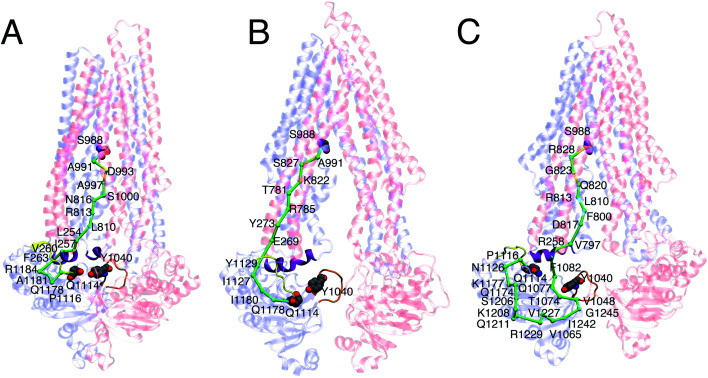
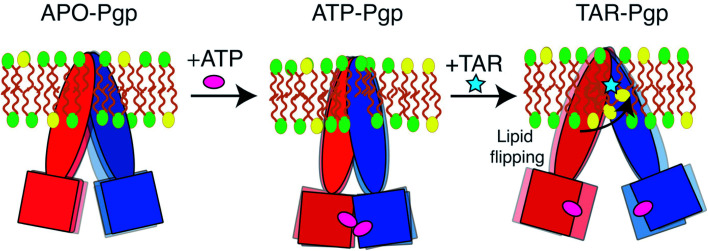
P-glycoprotein (Pgp) is a major efflux pump in humans, overexpressed in a variety of cancers and associated with the development of multi-drug resistance. Allosteric modulation by various ligands (e.g., transport substrates, inhibitors, and ATP) has been biochemically shown to directly influence structural dynamics, and thereby, the function of Pgp. However, the molecular details of such effects, particularly with respect to the role and involvement of the surrounding lipids, are not well established. Here, we employ all-atom molecular dynamics (MD) simulations to study the conformational landscape of Pgp in the presence of a high-affinity, third-generation inhibitor, tariquidar, in comparison to the nucleotide-free (APO) and the ATP-bound states, in order to characterize the mechanical effects of the inhibitor that might be of relevance to its blocking mechanism of Pgp. Simulations in a multi-component lipid bilayer show a dynamic equilibrium between open(er) and more closed inward-facing (IF) conformations in the APO state, with binding of ATP shifting the equilibrium towards conformations more prone to ATP hydrolysis and subsequent events in the transport cycle. In the presence of the inhibitor bound to the drug-binding pocket within the transmembrane domain (TMD), Pgp samples more open IF conformations, and the nucleotide binding domains (NBDs) become highly dynamic. Interestingly, and reproduced in multiple independent simulations, the inhibitor is observed to facilitate recruitment of lipid molecules into the Pgp lumen through the two proposed drug-entry portals, where the lipid head groups from the cytoplasmic leaflet penetrate into and, in some cases, translocate inside the TMD, while the lipid tails remain extended into the bulk lipid environment. These "wedge" lipids likely enhance the inhibitor-induced conformational restriction of the TMD leading to the differential modulation of coupling pathways observed with the NBDs downstream. We suggest a novel inhibitory mechanism for tariquidar, and potentially for related third-generation Pgp inhibitors, where lipids are seen to enhance the inhibitory role in the catalytic cycle of membrane transporters.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
The authors declare no conflict of interests.
Figures
Similar articles
-
Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer-associated P-glycoprotein during ATP hydrolysis.J Biol Chem. 2017 Dec 15;292(50):20412-20424. doi: 10.1074/jbc.M117.814186. Epub 2017 Oct 9. J Biol Chem. 2017. PMID: 29018094 Free PMC article.
-
On the origin of large flexibility of P-glycoprotein in the inward-facing state.J Biol Chem. 2013 Jun 28;288(26):19211-20. doi: 10.1074/jbc.M113.450114. Epub 2013 May 8. J Biol Chem. 2013. PMID: 23658020 Free PMC article.
-
Membrane-assisted tariquidar access and binding mechanisms of human ATP-binding cassette transporter P-glycoprotein.Front Mol Biosci. 2024 Mar 15;11:1364494. doi: 10.3389/fmolb.2024.1364494. eCollection 2024. Front Mol Biosci. 2024. PMID: 38560519 Free PMC article.
-
Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function.Front Oncol. 2014 Mar 3;4:41. doi: 10.3389/fonc.2014.00041. eCollection 2014. Front Oncol. 2014. PMID: 24624364 Free PMC article. Review.
-
About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work.Mol Cancer Ther. 2007 Jan;6(1):13-23. doi: 10.1158/1535-7163.MCT-06-0155. Mol Cancer Ther. 2007. PMID: 17237262 Review.
Cited by
-
Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein.Life (Basel). 2022 Jun 15;12(6):897. doi: 10.3390/life12060897. Life (Basel). 2022. PMID: 35743927 Free PMC article. Review.
-
Druggable Lipid Binding Sites in Pentameric Ligand-Gated Ion Channels and Transient Receptor Potential Channels.Front Physiol. 2022 Jan 4;12:798102. doi: 10.3389/fphys.2021.798102. eCollection 2021. Front Physiol. 2022. PMID: 35069257 Free PMC article. Review.
-
Cholesterol-Depletion-Induced Membrane Repair Carries a Raft Conformer of P-Glycoprotein to the Cell Surface, Indicating Enhanced Cholesterol Trafficking in MDR Cells, Which Makes Them Resistant to Cholesterol Modifications.Int J Mol Sci. 2023 Aug 2;24(15):12335. doi: 10.3390/ijms241512335. Int J Mol Sci. 2023. PMID: 37569709 Free PMC article.
-
Probing the Allosteric Modulation of P-Glycoprotein: A Medicinal Chemistry Approach Toward the Identification of Noncompetitive P-Gp Inhibitors.ACS Omega. 2023 Mar 14;8(12):11281-11287. doi: 10.1021/acsomega.2c08273. eCollection 2023 Mar 28. ACS Omega. 2023. PMID: 37008154 Free PMC article.
-
On the interplay between lipids and asymmetric dynamics of an NBS degenerate ABC transporter.Commun Biol. 2023 Feb 3;6(1):149. doi: 10.1038/s42003-023-04537-3. Commun Biol. 2023. PMID: 36737455 Free PMC article.
References
-
- Dean M. Hamon Y. Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001;42:1007–1017. - PubMed
-
- Locher K. P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016;23:487. - PubMed
-
- Busch W. Saier M. H. The transporter classification (TC) system. Crit. Rev. Biochem. Mol. Biol. 2002;37:287–337. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous