

Whole-Exome Sequencing Reveals Rare Germline Mutations in Patients With Hemifacial Microsomia
- PMID: 34079577
- PMCID: PMC8165440
- DOI: 10.3389/fgene.2021.580761
Whole-Exome Sequencing Reveals Rare Germline Mutations in Patients With Hemifacial Microsomia
Abstract
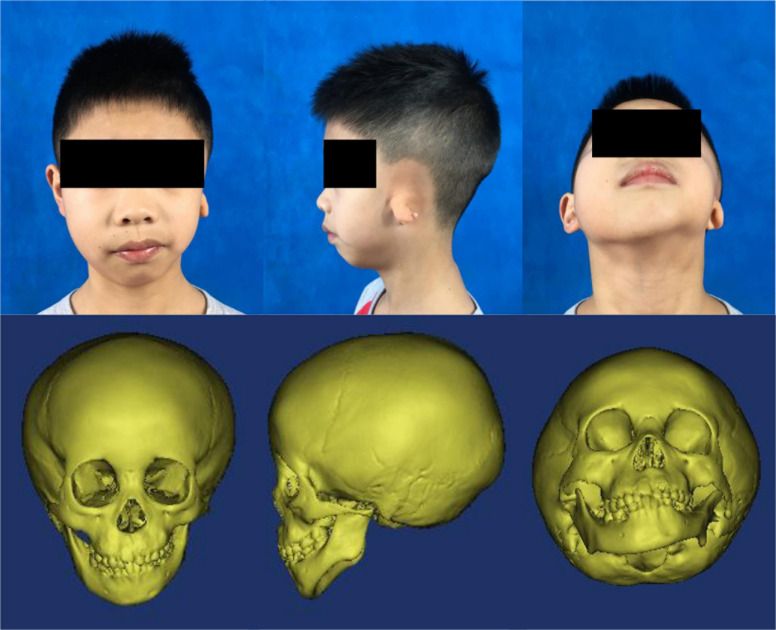
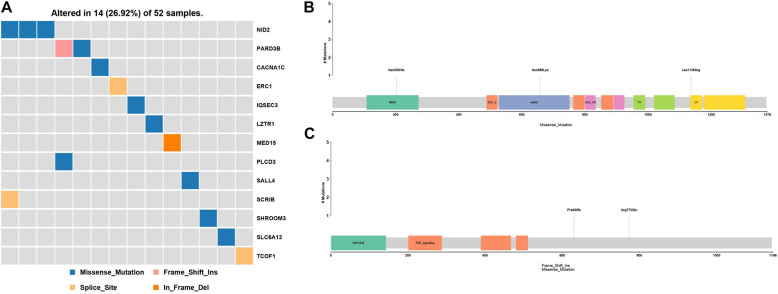
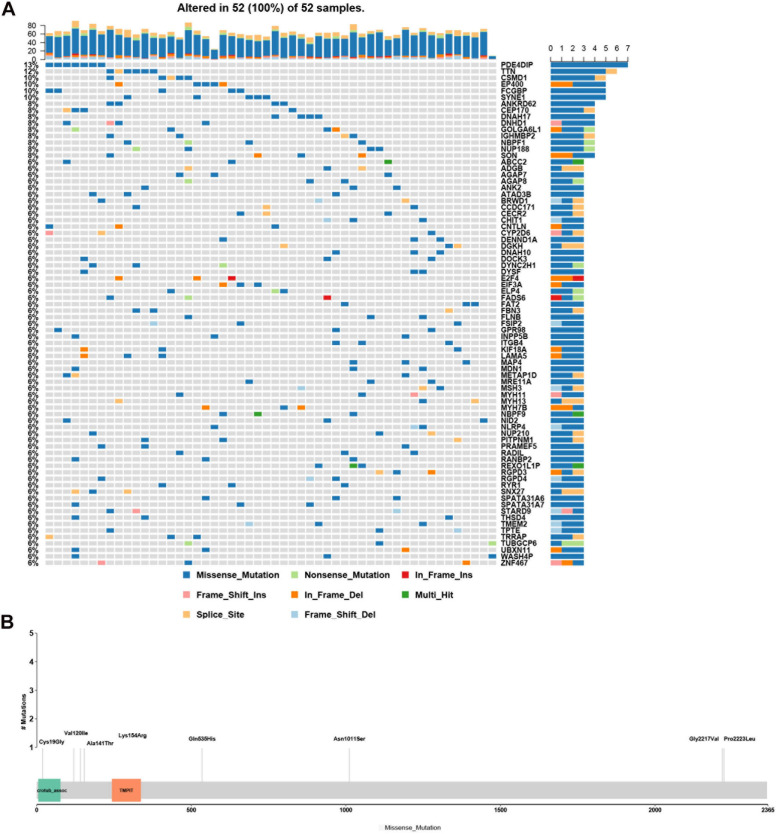
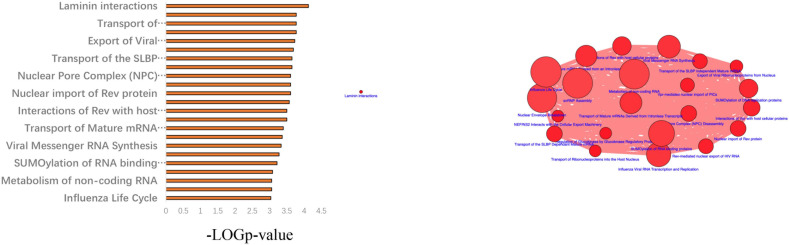
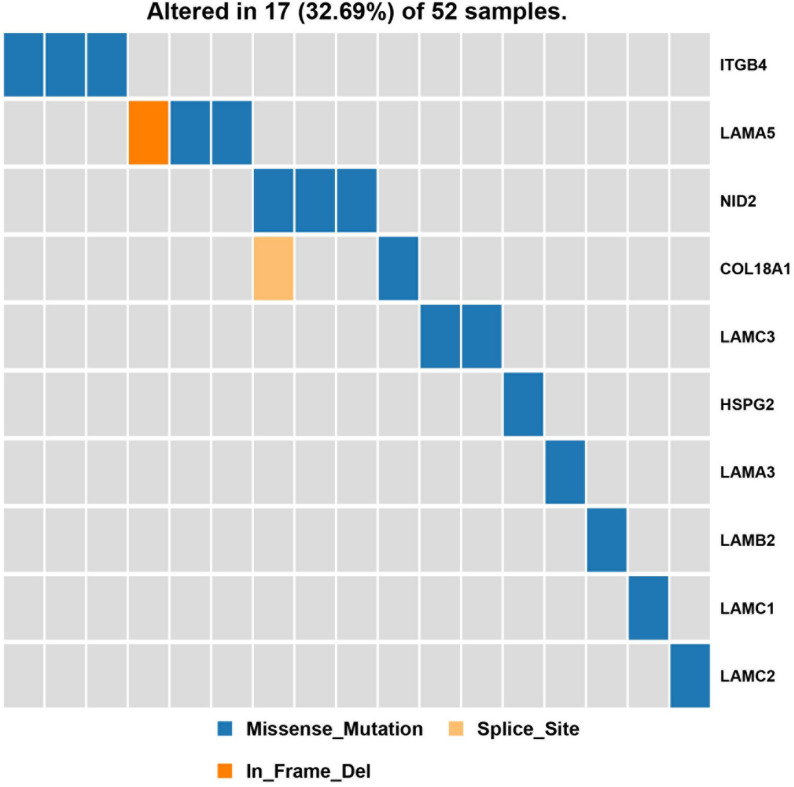
Hemifacial microsomia (HFM) is a rare congenital disease characterized by a spectrum of craniomaxillofacial malformations, including unilateral hypoplasia of the mandible and surrounding structures. Genetic predisposition for HFM is evident but the causative genes have not been fully understood. Thus, in the present study, we used whole-exome sequencing to screen 52 patients with HFM for rare germline mutations. We revealed 3,341 rare germline mutations in this patient cohort, including those in 13 genes previously shown to be associated with HFM. Among these HFM-related genes, NID2 was most frequently mutated (in 3/52 patients). PED4DIP, which has not been previously associated with HFM, exhibited rare variants most frequently (in 7/52 patients). Pathway enrichment analysis of genes that were mutated in >2 patients predicted the "laminin interactions" pathway to be most significantly disrupted, predominantly by mutations in ITGB4, NID2, or LAMA5. In summary, this study is the first to identify rare germline mutations in HFM. The likely disruptions in the signaling pathways due to the mutations reported here may be considered potential causes of HFM.
Keywords: hemifacial microsomia; mandibular hypoplasia; pathway enrichment analysis; rare germline mutations; whole-exome sequencing.
Copyright © 2021 Chen, Liu, Mar Aung, Zhang and Chai.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Whole-exome sequencing for monozygotic twins discordant for hemifacial microsomia.J Craniomaxillofac Surg. 2018 May;46(5):802-807. doi: 10.1016/j.jcms.2018.02.005. Epub 2018 Feb 16. J Craniomaxillofac Surg. 2018. PMID: 29551253
-
Transcriptome sequencing of facial adipose tissue reveals alterations in mRNAs of hemifacial microsomia.Front Pediatr. 2023 Feb 13;11:1099841. doi: 10.3389/fped.2023.1099841. eCollection 2023. Front Pediatr. 2023. PMID: 36861077 Free PMC article.
-
Complexities of Hemifacial Microsomia: A Case Study of Mandibular Hypoplasia and Ear Deformity.Cureus. 2024 Jul 13;16(7):e64499. doi: 10.7759/cureus.64499. eCollection 2024 Jul. Cureus. 2024. PMID: 39139347 Free PMC article.
-
Etiology and Pathogenesis of Hemifacial Microsomia.J Dent Res. 2018 Nov;97(12):1297-1305. doi: 10.1177/0022034518795609. Epub 2018 Sep 11. J Dent Res. 2018. PMID: 30205013 Review.
-
Interrelationships of the hemifacial microsomia-VATER, VATER, and sirenomelia phenotypes.Am J Med Genet. 1993 Aug 1;47(1):75-84. doi: 10.1002/ajmg.1320470116. Am J Med Genet. 1993. PMID: 8368258 Review.
Cited by
-
The Enigmatic Etiology of Oculo-Auriculo-Vertebral Spectrum (OAVS): An Exploratory Gene Variant Interaction Approach in Candidate Genes.Life (Basel). 2022 Oct 28;12(11):1723. doi: 10.3390/life12111723. Life (Basel). 2022. PMID: 36362878 Free PMC article.
-
Identification of novel TMEM231 gene splice variants and pathological findings in a fetus with Meckel Syndrome.Front Genet. 2023 Sep 6;14:1252873. doi: 10.3389/fgene.2023.1252873. eCollection 2023. Front Genet. 2023. PMID: 37736303 Free PMC article.
References
-
- Abul-Husn N. S., Manickam K., Jones L. K., Wright E. A., Hartzel D. N., Gonzaga-Jauregui C., et al. (2016). Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 354:aaf7000. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous