

DDX17 is involved in DNA damage repair and modifies FUS toxicity in an RGG-domain dependent manner
- PMID: 34061233
- PMCID: PMC8856901
- DOI: 10.1007/s00401-021-02333-z
DDX17 is involved in DNA damage repair and modifies FUS toxicity in an RGG-domain dependent manner
Abstract
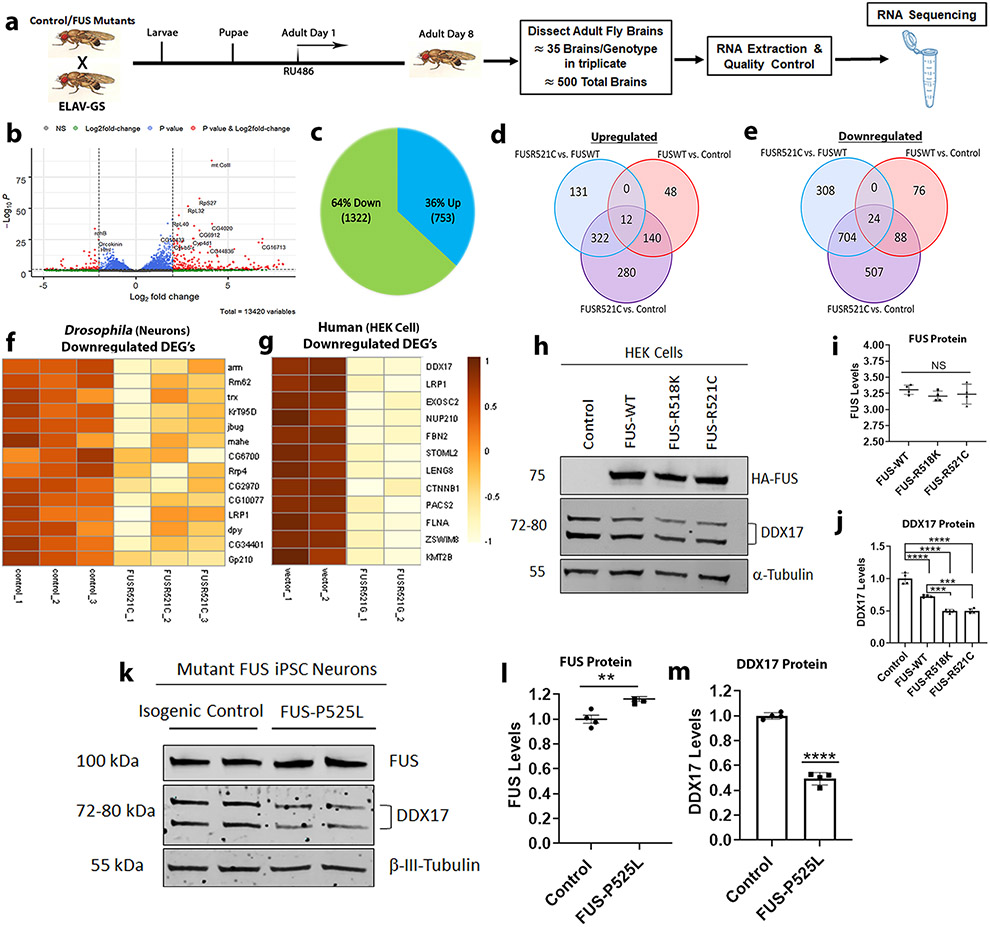
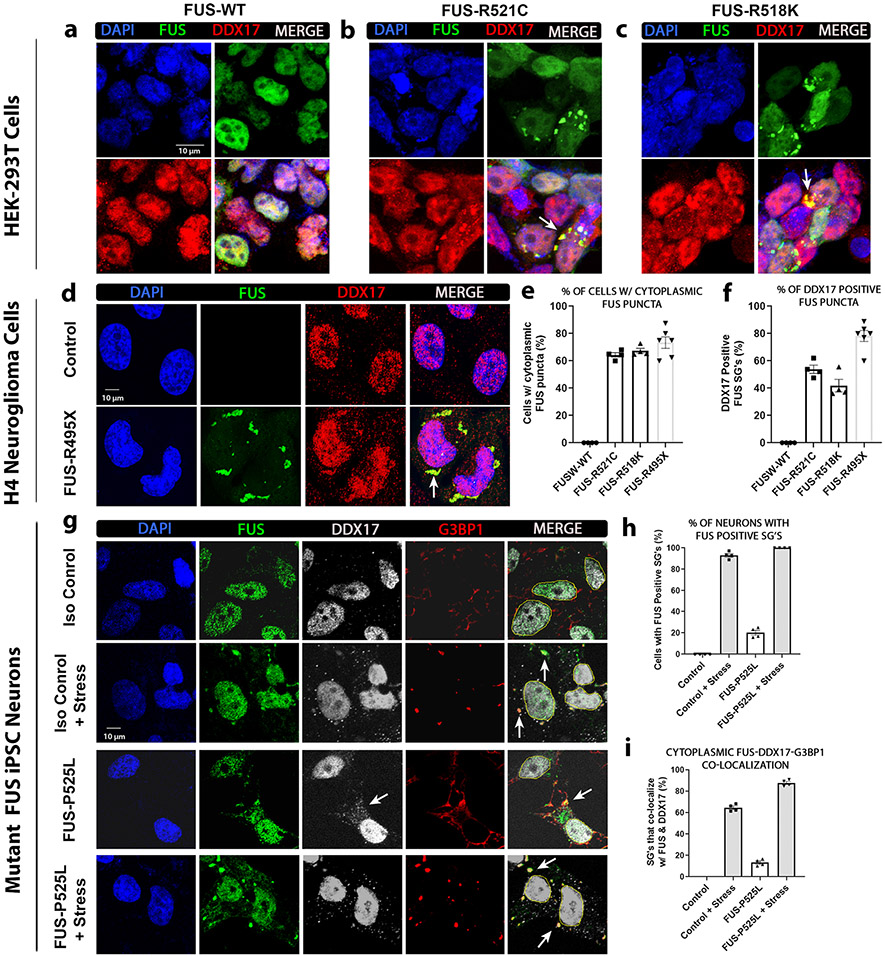
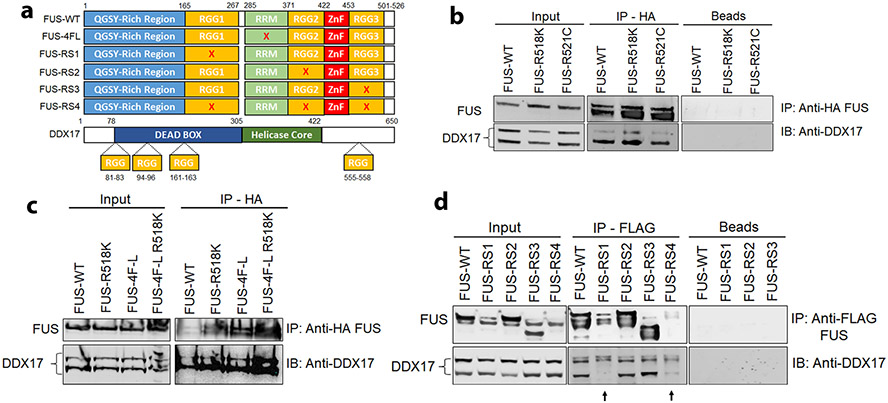
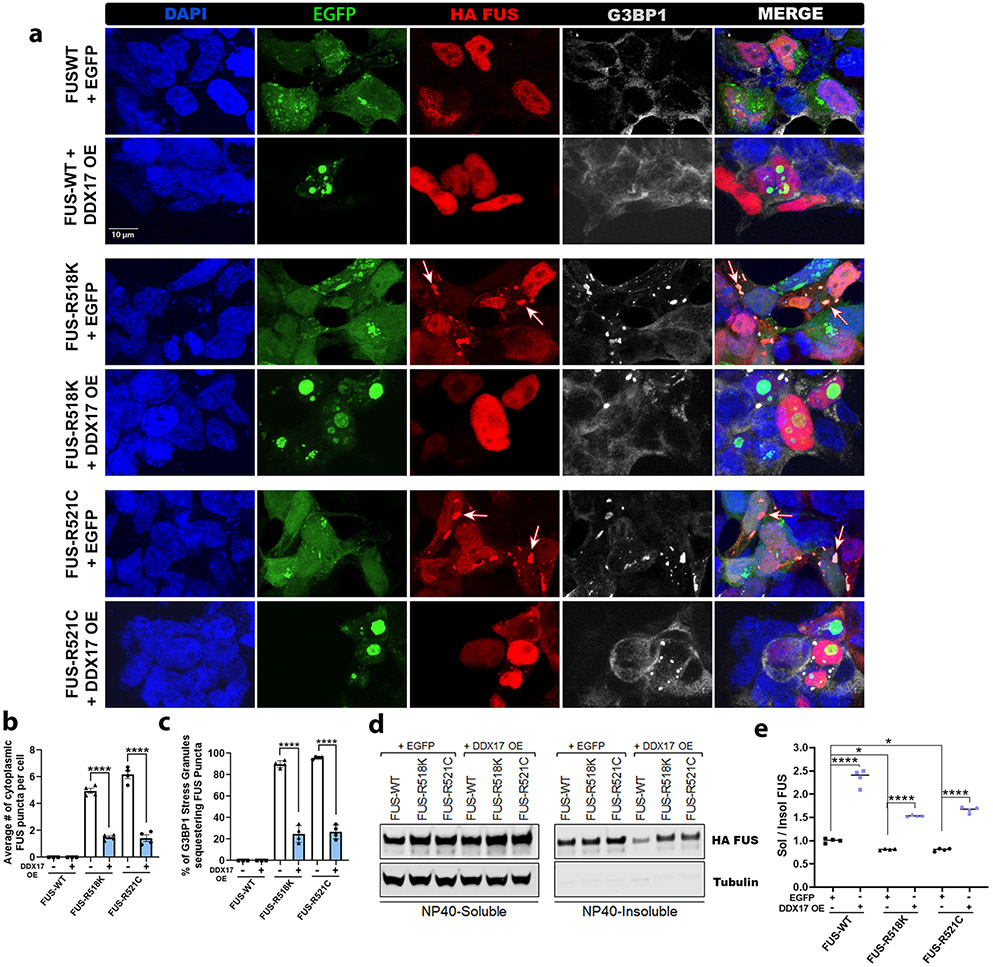
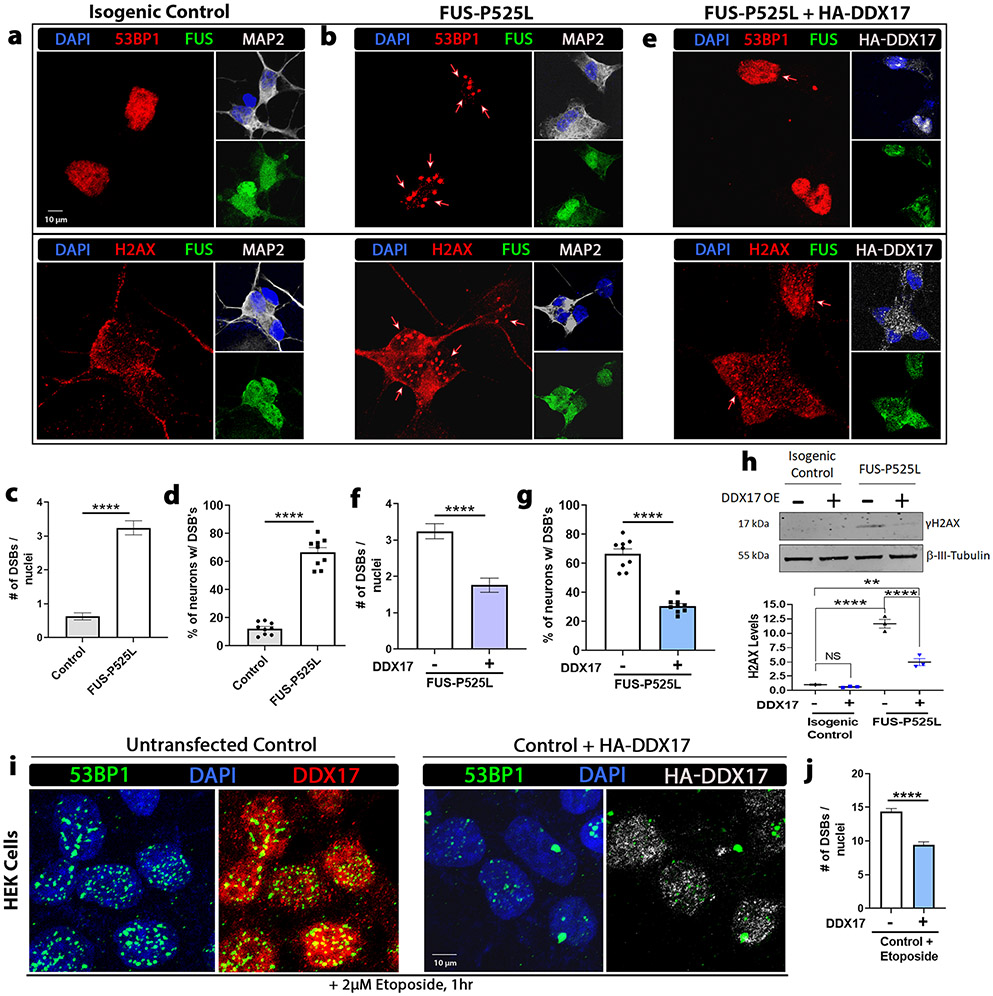
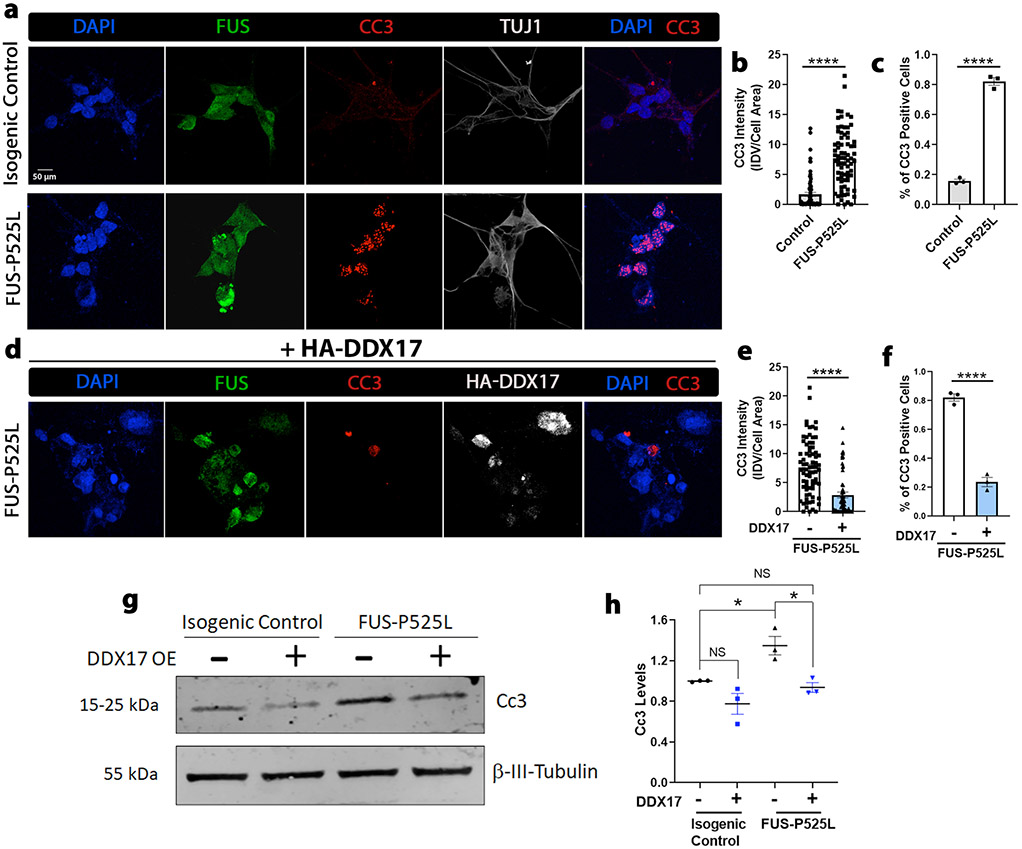
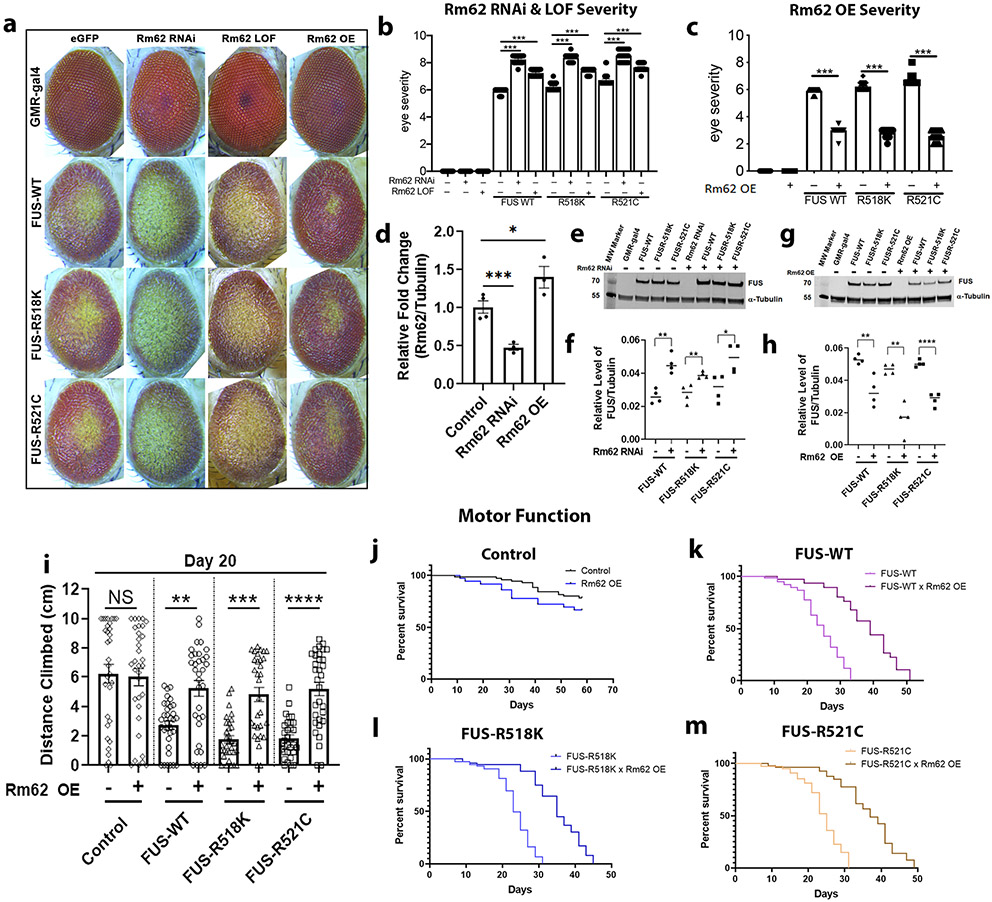
Mutations in the RNA binding protein, Fused in Sarcoma (FUS), lead to amyotrophic lateral sclerosis (ALS), the most frequent form of motor neuron disease. Cytoplasmic aggregation and defective DNA repair machinery are etiologically linked to mutant FUS-associated ALS. Although FUS is involved in numerous aspects of RNA processing, little is understood about the pathophysiological mechanisms of mutant FUS. Here, we employed RNA-sequencing technology in Drosophila brains expressing FUS to identify significantly altered genes and pathways involved in FUS-mediated neurodegeneration. We observed the expression levels of DEAD-Box Helicase 17 (DDX17) to be significantly downregulated in response to mutant FUS in Drosophila and human cell lines. Mutant FUS recruits nuclear DDX17 into cytoplasmic stress granules and physically interacts with DDX17 through the RGG1 domain of FUS. Ectopic expression of DDX17 reduces cytoplasmic mislocalization and sequestration of mutant FUS into cytoplasmic stress granules. We identified DDX17 as a novel regulator of the DNA damage response pathway whose upregulation repairs defective DNA damage repair machinery caused by mutant neuronal FUS ALS. In addition, we show DDX17 is a novel modifier of FUS-mediated neurodegeneration in vivo. Our findings indicate DDX17 is downregulated in response to mutant FUS, and restoration of DDX17 levels suppresses FUS-mediated neuropathogenesis and toxicity in vivo.
Keywords: ALS/FTD; DDX17; DNA-damage repair; Drosophila; FUS; Motor neuron disease; Neurodegeneration; RGG-domain; iPSC neurons.
© 2021. The Author(s), under exclusive licence to Springer-Verlag GmbH Germany, part of Springer Nature.
Figures
Similar articles
-
RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations.Hum Mol Genet. 2013 Mar 15;22(6):1193-205. doi: 10.1093/hmg/dds526. Epub 2012 Dec 20. Hum Mol Genet. 2013. PMID: 23257289 Free PMC article.
-
Muscleblind acts as a modifier of FUS toxicity by modulating stress granule dynamics and SMN localization.Nat Commun. 2019 Dec 6;10(1):5583. doi: 10.1038/s41467-019-13383-z. Nat Commun. 2019. PMID: 31811140 Free PMC article.
-
Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity.Acta Neuropathol. 2016 Apr;131(4):605-20. doi: 10.1007/s00401-015-1530-0. Epub 2016 Jan 4. Acta Neuropathol. 2016. PMID: 26728149 Free PMC article.
-
Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis.Brain Res. 2016 Sep 15;1647:65-78. doi: 10.1016/j.brainres.2016.03.036. Epub 2016 Mar 28. Brain Res. 2016. PMID: 27033831 Free PMC article. Review.
-
Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS.Neurology. 2011 Oct 25;77(17):1636-43. doi: 10.1212/WNL.0b013e3182343365. Epub 2011 Sep 28. Neurology. 2011. PMID: 21956718 Free PMC article. Review.
Cited by
-
Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers.Cell Mol Life Sci. 2021 Sep;78(17-18):6143-6160. doi: 10.1007/s00018-021-03905-8. Epub 2021 Jul 28. Cell Mol Life Sci. 2021. PMID: 34322715 Free PMC article. Review.
-
Mitochondrial dynamics and metabolism across skin cells: implications for skin homeostasis and aging.Front Physiol. 2023 Nov 15;14:1284410. doi: 10.3389/fphys.2023.1284410. eCollection 2023. Front Physiol. 2023. PMID: 38046945 Free PMC article. Review.
-
Drosha-dependent microRNAs modulate FUS-mediated neurodegeneration in vivo.Nucleic Acids Res. 2023 Nov 10;51(20):11258-11276. doi: 10.1093/nar/gkad774. Nucleic Acids Res. 2023. PMID: 37791873 Free PMC article.
-
C9orf72 poly(PR) mediated neurodegeneration is associated with nucleolar stress.iScience. 2023 Jul 28;26(9):107505. doi: 10.1016/j.isci.2023.107505. eCollection 2023 Sep 15. iScience. 2023. PMID: 37664610 Free PMC article.
-
Lactobacillus Eats Amyloid Plaque and Post-Biotically Attenuates Senescence Due to Repeat Expansion Disorder and Alzheimer's Disease.Antioxidants (Basel). 2024 Oct 12;13(10):1225. doi: 10.3390/antiox13101225. Antioxidants (Basel). 2024. PMID: 39456478 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
