

Impact of DNA methylation on 3D genome structure
- PMID: 34050148
- PMCID: PMC8163762
- DOI: 10.1038/s41467-021-23142-8
Impact of DNA methylation on 3D genome structure
Abstract
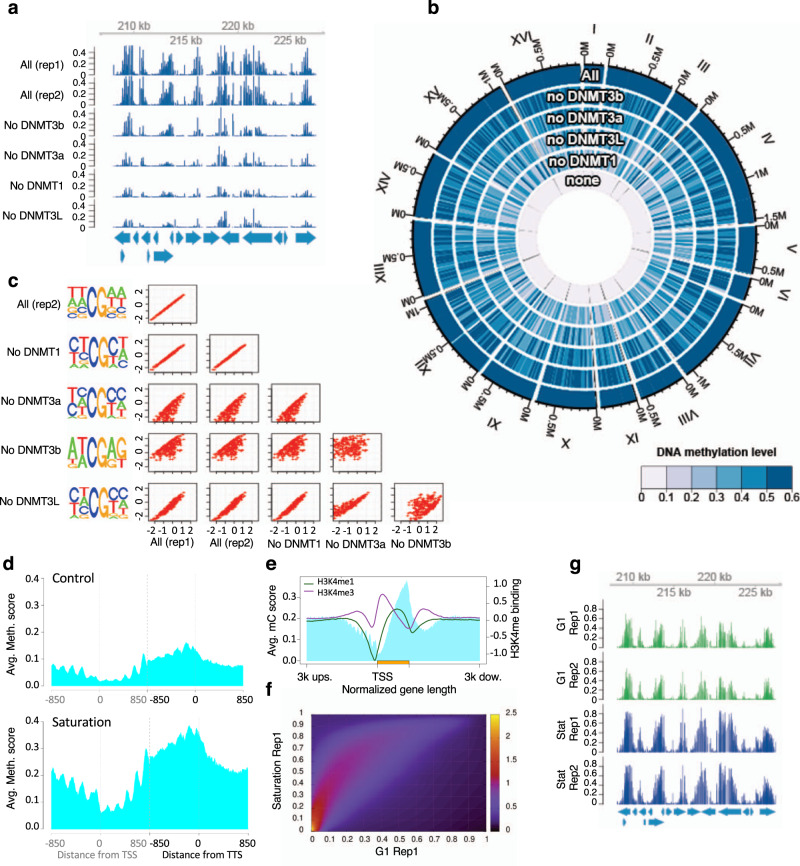
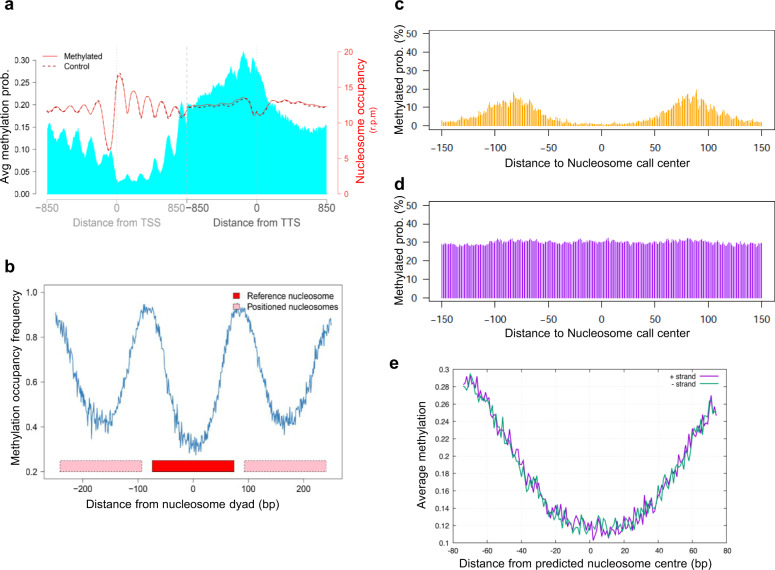
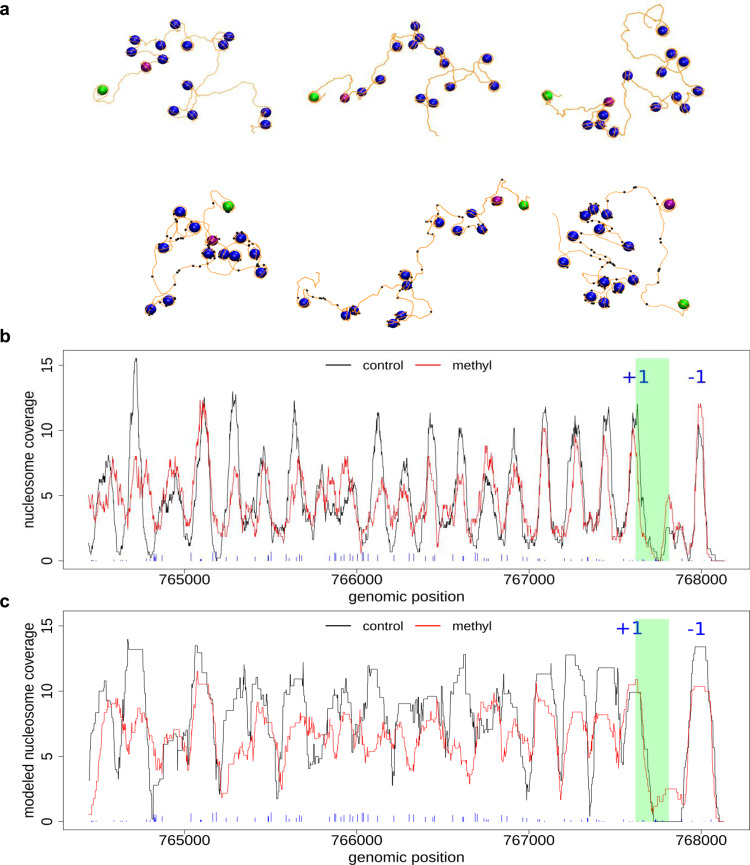
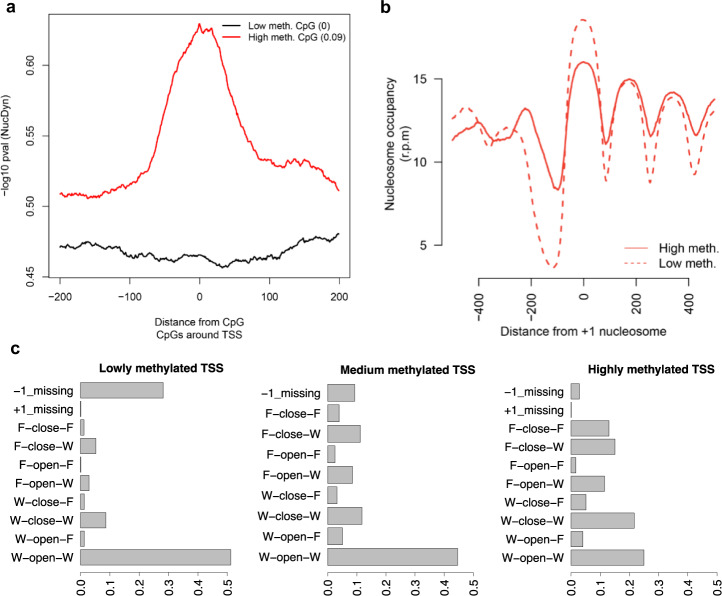
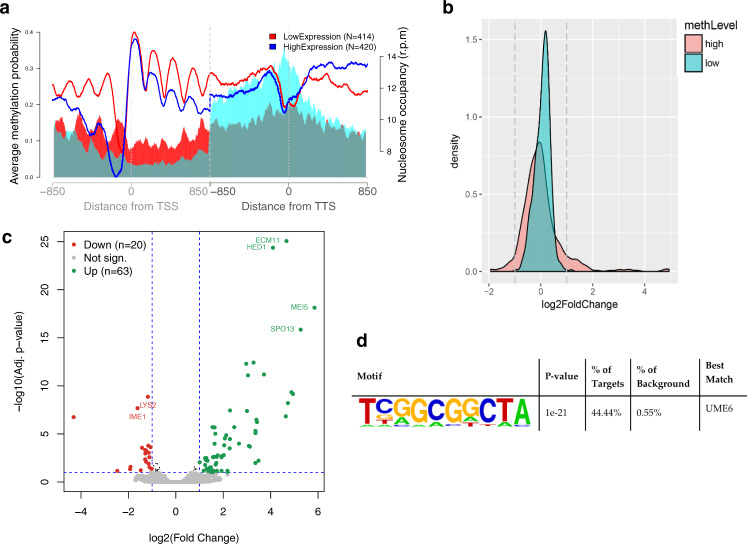
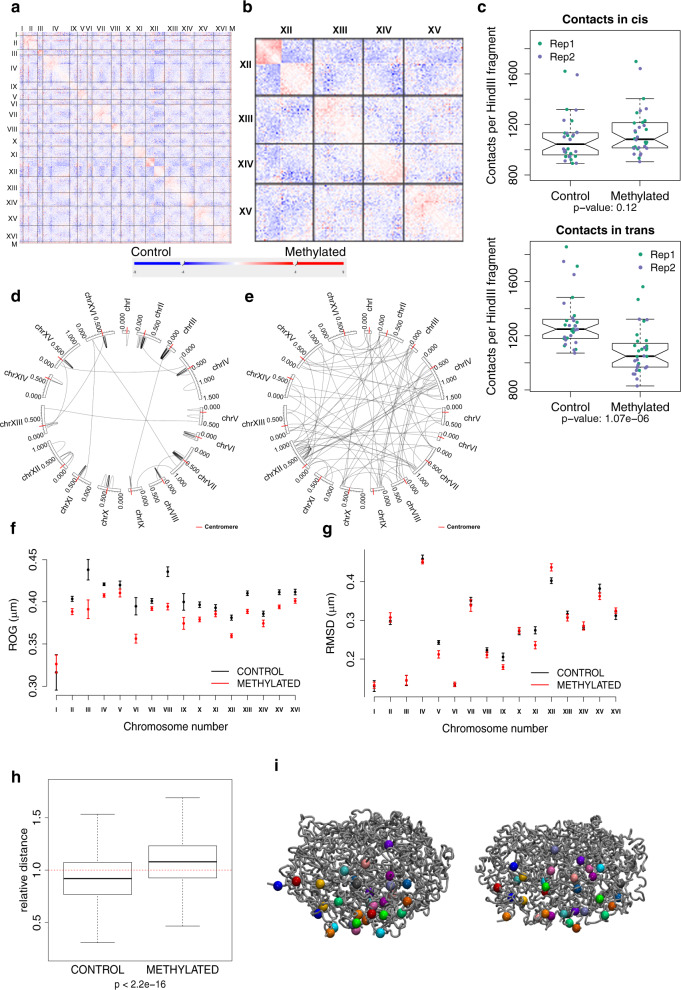
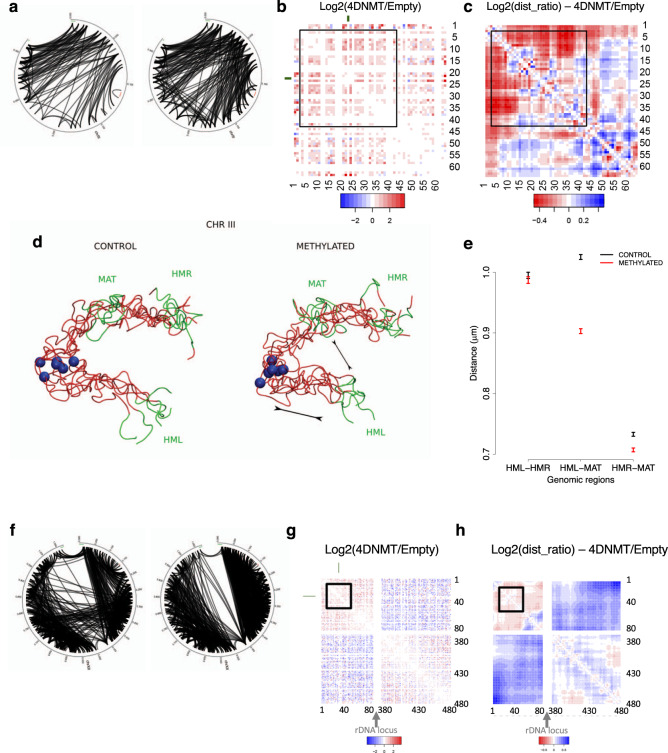
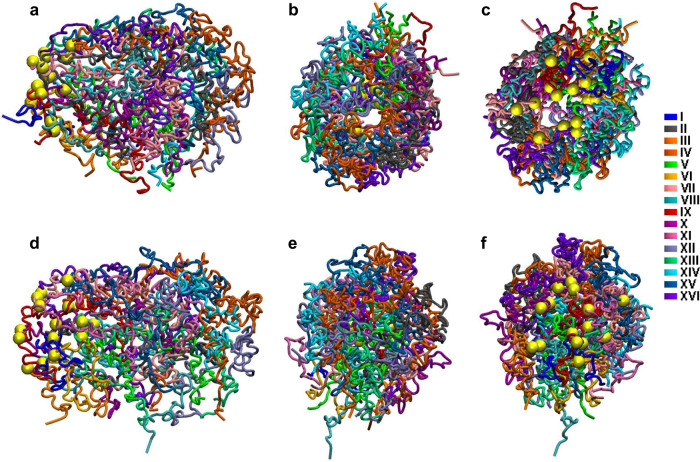
Determining the effect of DNA methylation on chromatin structure and function in higher organisms is challenging due to the extreme complexity of epigenetic regulation. We studied a simpler model system, budding yeast, that lacks DNA methylation machinery making it a perfect model system to study the intrinsic role of DNA methylation in chromatin structure and function. We expressed the murine DNA methyltransferases in Saccharomyces cerevisiae and analyzed the correlation between DNA methylation, nucleosome positioning, gene expression and 3D genome organization. Despite lacking the machinery for positioning and reading methylation marks, induced DNA methylation follows a conserved pattern with low methylation levels at the 5' end of the gene increasing gradually toward the 3' end, with concentration of methylated DNA in linkers and nucleosome free regions, and with actively expressed genes showing low and high levels of methylation at transcription start and terminating sites respectively, mimicking the patterns seen in mammals. We also see that DNA methylation increases chromatin condensation in peri-centromeric regions, decreases overall DNA flexibility, and favors the heterochromatin state. Taken together, these results demonstrate that methylation intrinsically modulates chromatin structure and function even in the absence of cellular machinery evolved to recognize and process the methylation signal.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Ruler elements in chromatin remodelers set nucleosome array spacing and phasing.Nat Commun. 2021 May 28;12(1):3232. doi: 10.1038/s41467-021-23015-0. Nat Commun. 2021. PMID: 34050140 Free PMC article.
-
The N-terminus of histone H3 is required for de novo DNA methylation in chromatin.Proc Natl Acad Sci U S A. 2009 Dec 29;106(52):22187-92. doi: 10.1073/pnas.0905767106. Epub 2009 Dec 14. Proc Natl Acad Sci U S A. 2009. PMID: 20018712 Free PMC article.
-
Genome information processing by the INO80 chromatin remodeler positions nucleosomes.Nat Commun. 2021 May 28;12(1):3231. doi: 10.1038/s41467-021-23016-z. Nat Commun. 2021. PMID: 34050142 Free PMC article.
-
Unbelievable but True: Epigenetics and Chromatin in Fungi.Trends Genet. 2021 Jan;37(1):12-20. doi: 10.1016/j.tig.2020.09.016. Epub 2020 Oct 19. Trends Genet. 2021. PMID: 33092902 Free PMC article. Review.
-
Chromatin regulation of plant development.Curr Opin Plant Biol. 2003 Feb;6(1):20-8. doi: 10.1016/s1369526602000079. Curr Opin Plant Biol. 2003. PMID: 12495747 Review.
Cited by
-
The Diversity of Methylation Patterns in Serous Borderline Ovarian Tumors and Serous Ovarian Carcinomas.Cancers (Basel). 2024 Oct 18;16(20):3524. doi: 10.3390/cancers16203524. Cancers (Basel). 2024. PMID: 39456618 Free PMC article.
-
Understanding the molecular mechanisms of human diseases: the benefits of fission yeasts.Microb Cell. 2024 Aug 2;11:288-311. doi: 10.15698/mic2024.08.833. eCollection 2024. Microb Cell. 2024. PMID: 39104724 Free PMC article.
-
A commentary on the ABC consortium and its impact on the development of mesoscopic models of DNA.Biophys Rev. 2024 May 7;16(3):273-274. doi: 10.1007/s12551-024-01196-4. eCollection 2024 Jun. Biophys Rev. 2024. PMID: 39099835
-
MD-DATA: the legacy of the ABC Consortium.Biophys Rev. 2024 May 7;16(3):269-271. doi: 10.1007/s12551-024-01197-3. eCollection 2024 Jun. Biophys Rev. 2024. PMID: 39099843 Free PMC article.
-
DNA methylation cues in nucleosome geometry, stability and unwrapping.Nucleic Acids Res. 2022 Feb 28;50(4):1864-1874. doi: 10.1093/nar/gkac097. Nucleic Acids Res. 2022. PMID: 35166834 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
