

Interplay between inflammation and thrombosis in cardiovascular pathology
- PMID: 33958774
- PMCID: PMC8100938
- DOI: 10.1038/s41569-021-00552-1
Interplay between inflammation and thrombosis in cardiovascular pathology
Abstract
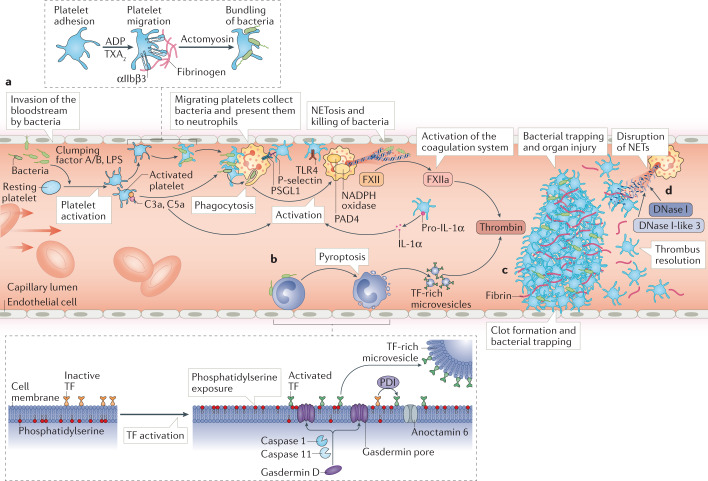
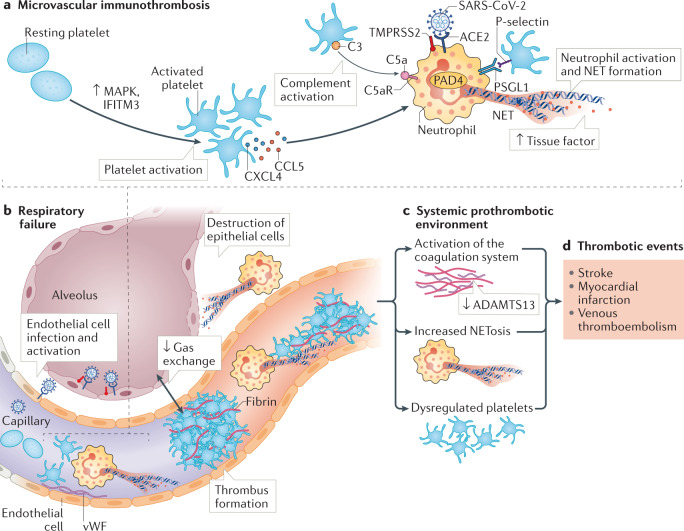
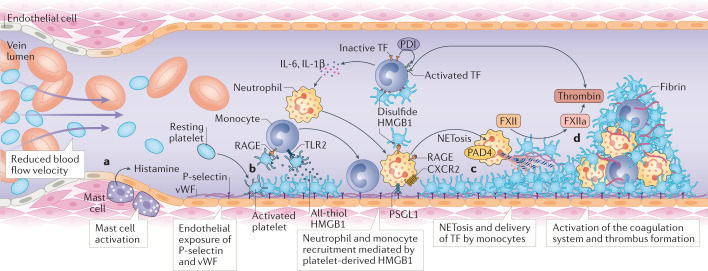
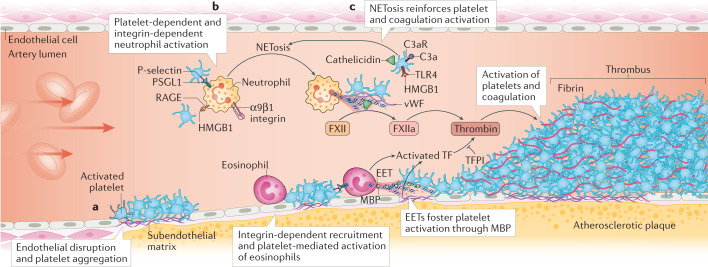
Thrombosis is the most feared complication of cardiovascular diseases and a main cause of death worldwide, making it a major health-care challenge. Platelets and the coagulation cascade are effectively targeted by antithrombotic approaches, which carry an inherent risk of bleeding. Moreover, antithrombotics cannot completely prevent thrombotic events, implicating a therapeutic gap due to a third, not yet adequately addressed mechanism, namely inflammation. In this Review, we discuss how the synergy between inflammation and thrombosis drives thrombotic diseases. We focus on the huge potential of anti-inflammatory strategies to target cardiovascular pathologies. Findings in the past decade have uncovered a sophisticated connection between innate immunity, platelet activation and coagulation, termed immunothrombosis. Immunothrombosis is an important host defence mechanism to limit systemic spreading of pathogens through the bloodstream. However, the aberrant activation of immunothrombosis in cardiovascular diseases causes myocardial infarction, stroke and venous thromboembolism. The clinical relevance of aberrant immunothrombosis, referred to as thromboinflammation, is supported by the increased risk of cardiovascular events in patients with inflammatory diseases but also during infections, including in COVID-19. Clinical trials in the past 4 years have confirmed the anti-ischaemic effects of anti-inflammatory strategies, backing the concept of a prothrombotic function of inflammation. Targeting inflammation to prevent thrombosis leaves haemostasis mainly unaffected, circumventing the risk of bleeding associated with current approaches. Considering the growing number of anti-inflammatory therapies, it is crucial to appreciate their potential in covering therapeutic gaps in cardiovascular diseases.
© 2021. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
The Role of CLEC-2 and Its Ligands in Thromboinflammation.Front Immunol. 2021 Jun 9;12:688643. doi: 10.3389/fimmu.2021.688643. eCollection 2021. Front Immunol. 2021. PMID: 34177942 Free PMC article. Review.
-
Inflammation and cardiovascular diseases: lessons from seminal clinical trials.Cardiovasc Res. 2021 Jan 21;117(2):411-422. doi: 10.1093/cvr/cvaa211. Cardiovasc Res. 2021. PMID: 32666079 Review.
-
Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: mechanistic insights and implications for anti-thrombotic treatment.Cardiovasc Res. 2021 Jul 27;117(9):2030-2044. doi: 10.1093/cvr/cvaa263. Cardiovasc Res. 2021. PMID: 32931586 Free PMC article. Review.
-
Factor XII as a Therapeutic Target in Thromboembolic and Inflammatory Diseases.Arterioscler Thromb Vasc Biol. 2017 Jan;37(1):13-20. doi: 10.1161/ATVBAHA.116.308595. Epub 2016 Nov 10. Arterioscler Thromb Vasc Biol. 2017. PMID: 27834692 Review.
-
Polyphosphate as modulator of hemostasis, thrombosis, and inflammation.J Thromb Haemost. 2015 Jun;13 Suppl 1(0 1):S92-7. doi: 10.1111/jth.12896. J Thromb Haemost. 2015. PMID: 26149055 Free PMC article. Review.
Cited by
-
Predictive models for thromboembolic events in giant cell arteritis: A US veterans health administration population-based study.Front Immunol. 2022 Nov 9;13:997347. doi: 10.3389/fimmu.2022.997347. eCollection 2022. Front Immunol. 2022. PMID: 36439172 Free PMC article.
-
Naoxintong capsule for treating cardiovascular and cerebrovascular diseases: from bench to bedside.Front Pharmacol. 2024 Jun 27;15:1402763. doi: 10.3389/fphar.2024.1402763. eCollection 2024. Front Pharmacol. 2024. PMID: 38994201 Free PMC article. Review.
-
The role of platelet mediated thromboinflammation in acute liver injury.Front Immunol. 2022 Oct 27;13:1037645. doi: 10.3389/fimmu.2022.1037645. eCollection 2022. Front Immunol. 2022. PMID: 36389830 Free PMC article. Review.
-
Lipidomics Revealed Plasma Phospholipid Profile Differences between Deceased and Recovered COVID-19 Patients.Biomolecules. 2022 Oct 15;12(10):1488. doi: 10.3390/biom12101488. Biomolecules. 2022. PMID: 36291697 Free PMC article.
-
Pathophysiological mechanisms of thrombosis in acute and long COVID-19.Front Immunol. 2022 Nov 16;13:992384. doi: 10.3389/fimmu.2022.992384. eCollection 2022. Front Immunol. 2022. PMID: 36466841 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
