

Identification of a novel inhibitor of SARS-CoV-2 3CL-PRO through virtual screening and molecular dynamics simulation
- PMID: 33954055
- PMCID: PMC8051358
- DOI: 10.7717/peerj.11261
Identification of a novel inhibitor of SARS-CoV-2 3CL-PRO through virtual screening and molecular dynamics simulation
Abstract
Background: The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has ravaged lives across the globe since December 2019, and new cases are still on the rise. Peoples' ongoing sufferings trigger scientists to develop safe and effective remedies to treat this deadly viral disease. While repurposing the existing FDA-approved drugs remains in the front line, exploring drug candidates from synthetic and natural compounds is also a viable alternative. This study employed a comprehensive computational approach to screen inhibitors for SARS-CoV-2 3CL-PRO (also known as the main protease), a prime molecular target to treat coronavirus diseases.
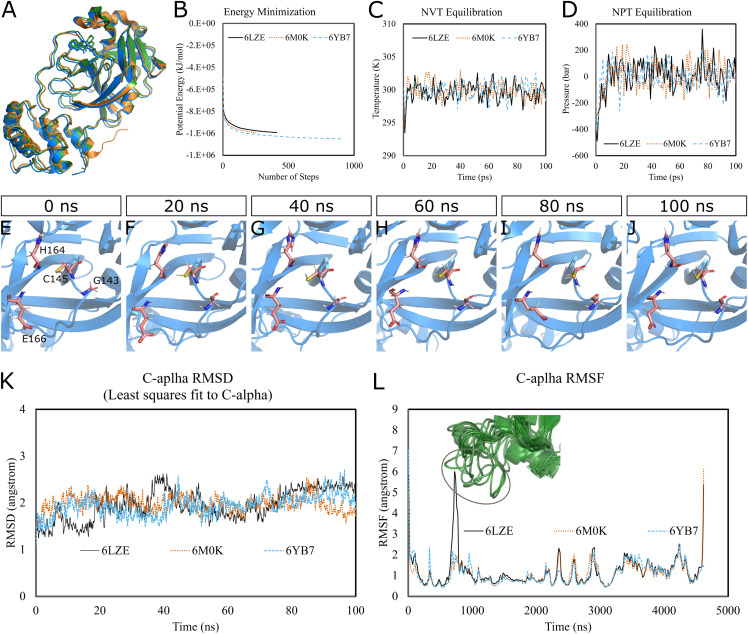
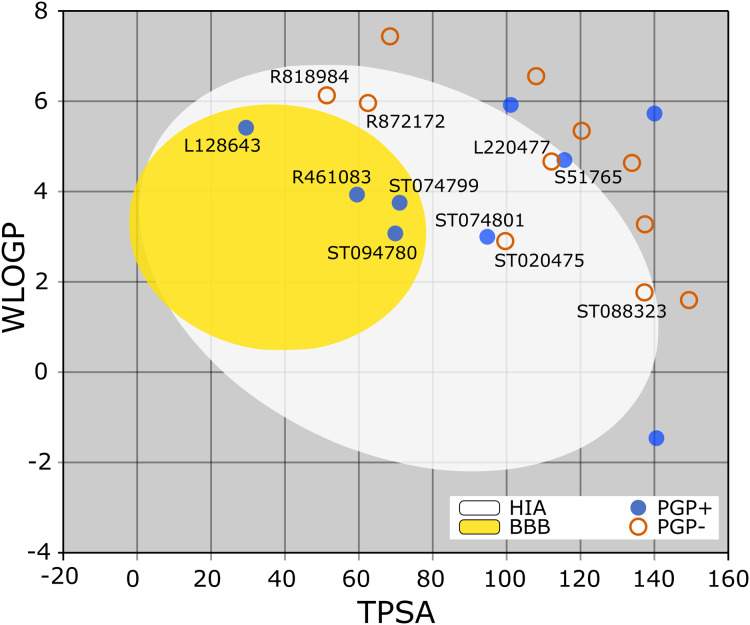
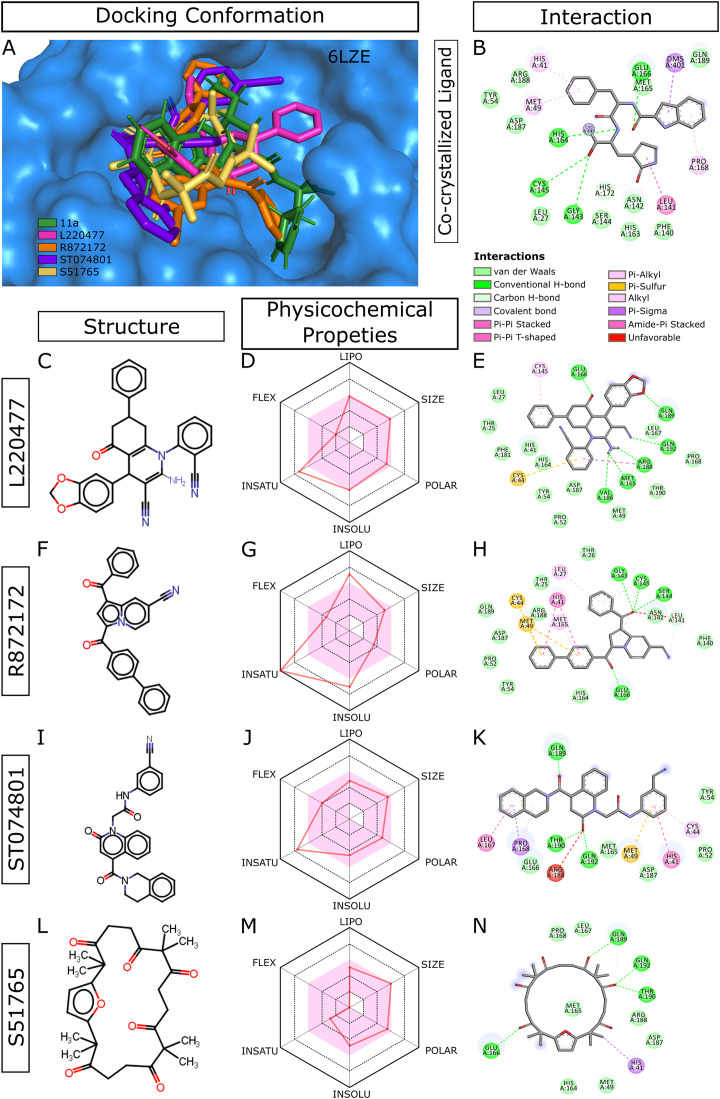
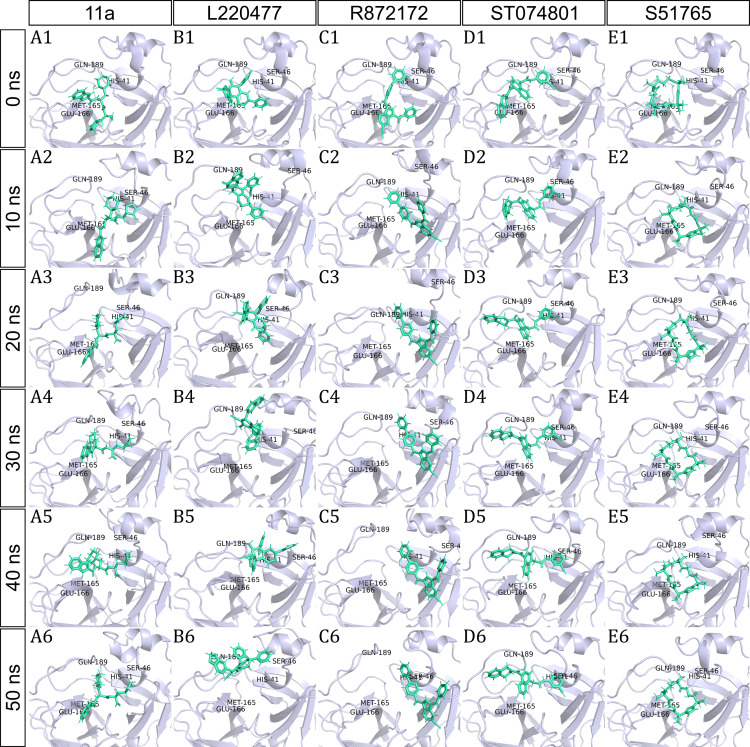
Methods: We performed 100 ns GROMACS molecular dynamics simulations of three high-resolution X-ray crystallographic structures of 3CL-PRO. We extracted frames at 10 ns intervals to mimic conformational diversities of the target protein in biological environments. We then used AutoDock Vina molecular docking to virtual screen the Sigma-Aldrich MyriaScreen Diversity Library II, a rich collection of 10,000 druglike small molecules with diverse chemotypes. Subsequently, we adopted in silico computation of physicochemical properties, pharmacokinetic parameters, and toxicity profiles. Finally, we analyzed hydrogen bonding and other protein-ligand interactions for the short-listed compounds.
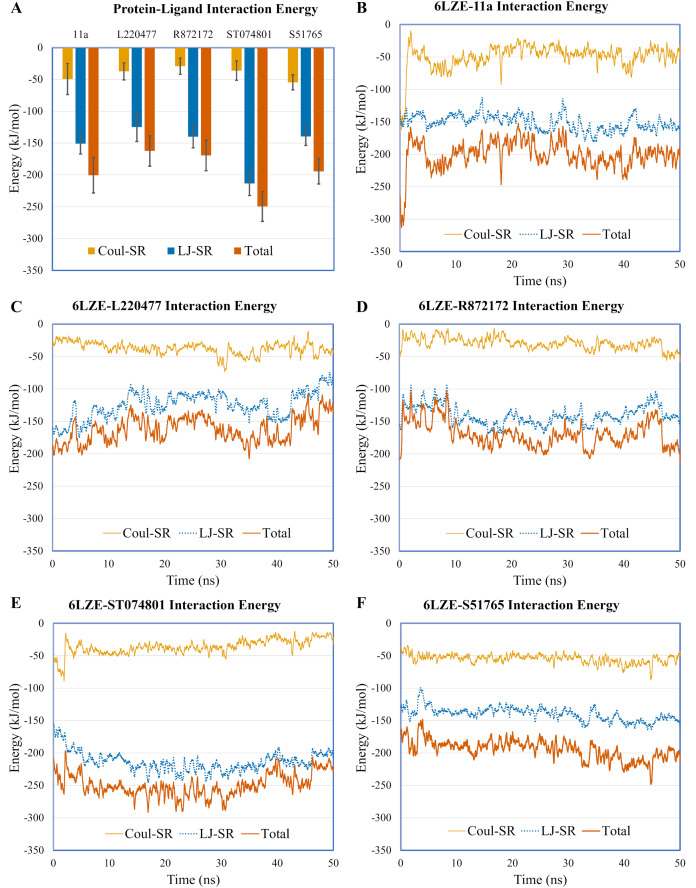
Results: Over the 100 ns molecular dynamics simulations of 3CL-PRO's crystal structures, 6LZE, 6M0K, and 6YB7, showed overall integrity with mean Cα root-mean-square deviation (RMSD) of 1.96 (±0.35) Å, 1.98 (±0.21) Å, and 1.94 (±0.25) Å, respectively. Average root-mean-square fluctuation (RMSF) values were 1.21 ± 0.79 (6LZE), 1.12 ± 0.72 (6M0K), and 1.11 ± 0.60 (6YB7). After two phases of AutoDock Vina virtual screening of the MyriaScreen Diversity Library II, we prepared a list of the top 20 ligands. We selected four promising leads considering predicted oral bioavailability, druglikeness, and toxicity profiles. These compounds also demonstrated favorable protein-ligand interactions. We then employed 50-ns molecular dynamics simulations for the four selected molecules and the reference ligand 11a in the crystallographic structure 6LZE. Analysis of RMSF, RMSD, and hydrogen bonding along the simulation trajectories indicated that S51765 would form a more stable protein-ligand complexe with 3CL-PRO compared to other molecules. Insights into short-range Coulombic and Lennard-Jones potentials also revealed favorable binding of S51765 with 3CL-PRO.
Conclusion: We identified a potential lead for antiviral drug discovery against the SARS-CoV-2 main protease. Our results will aid global efforts to find safe and effective remedies for COVID-19.
Keywords: 3CL-PRO; COVID-19; Coronavirus; Gromacs; Main protease; Mpro; SARS-CoV-2; Vina; docking; in silico.
© 2021 Bepari and Reza.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



Similar articles
-
Identifying the natural compound Catechin from tropical mangrove plants as a potential lead candidate against 3CLpro from SARS-CoV-2: An integrated in silico approach.J Biomol Struct Dyn. 2022;40(24):13392-13411. doi: 10.1080/07391102.2021.1988710. Epub 2021 Oct 13. J Biomol Struct Dyn. 2022. PMID: 34644249
-
Identification of novel inhibitors for SARS-CoV-2 as therapeutic options using machine learning-based virtual screening, molecular docking and MD simulation.Front Mol Biosci. 2023 Mar 7;10:1060076. doi: 10.3389/fmolb.2023.1060076. eCollection 2023. Front Mol Biosci. 2023. PMID: 36959979 Free PMC article.
-
Screening potential FDA-approved inhibitors of the SARS-CoV-2 major protease 3CLpro through high-throughput virtual screening and molecular dynamics simulation.Aging (Albany NY). 2021 Mar 7;13(5):6258-6272. doi: 10.18632/aging.202703. Epub 2021 Mar 7. Aging (Albany NY). 2021. PMID: 33678621 Free PMC article.
-
Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs.Precis Clin Med. 2021 Jan 18;4(1):1-16. doi: 10.1093/pcmedi/pbab001. eCollection 2021 Mar. Precis Clin Med. 2021. PMID: 33842834 Free PMC article. Review.
-
An understanding of coronavirus and exploring the molecular dynamics simulations to find promising candidates against the Mpro of nCoV to combat the COVID-19: A systematic review.J Infect Public Health. 2022 Nov;15(11):1326-1349. doi: 10.1016/j.jiph.2022.10.013. Epub 2022 Oct 19. J Infect Public Health. 2022. PMID: 36288640 Free PMC article. Review.
Cited by
-
Computational biophysics approach towards the discovery of multi-kinase blockers for the management of MAPK pathway dysregulation.Mol Divers. 2023 Oct;27(5):2093-2110. doi: 10.1007/s11030-022-10545-y. Epub 2022 Oct 19. Mol Divers. 2023. PMID: 36260173
-
Pharmacophore-based virtual screening of ZINC database, molecular modeling and designing new derivatives as potential HDAC6 inhibitors.Mol Divers. 2023 Oct;27(5):2053-2071. doi: 10.1007/s11030-022-10540-3. Epub 2022 Oct 10. Mol Divers. 2023. PMID: 36214962
-
Potential drugs against COVID-19 revealed by gene expression profile, molecular docking and molecular dynamic simulation.Future Virol. 2021 Jul:10.2217/fvl-2020-0392. doi: 10.2217/fvl-2020-0392. Epub 2021 Jul 20. Future Virol. 2021. PMID: 34306168 Free PMC article.
-
Identification of small molecules as potential inhibitors of interleukin 6: a multi-computational investigation.Mol Divers. 2023 Oct;27(5):2315-2330. doi: 10.1007/s11030-022-10558-7. Epub 2022 Nov 1. Mol Divers. 2023. PMID: 36319930 Free PMC article.
-
Quercetin and AMPK: A Dynamic Duo in Alleviating MG-Induced Inflammation via the AMPK/SIRT1/NF-κB Pathway.Molecules. 2023 Nov 2;28(21):7388. doi: 10.3390/molecules28217388. Molecules. 2023. PMID: 37959807 Free PMC article.
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Berendsen HJC, Van der Spoel D, Van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Computer Physics Communications. 1995;91(1–3):43–56. doi: 10.1016/0010-4655(95)00042-E. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
