

Sigma factor dependent translational activation in Bacillus subtilis
- PMID: 33927010
- PMCID: PMC8208050
- DOI: 10.1261/rna.078747.121
Sigma factor dependent translational activation in Bacillus subtilis
Abstract
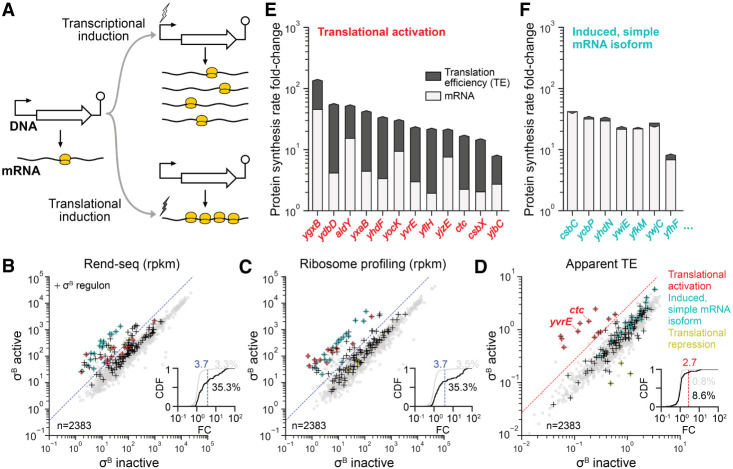
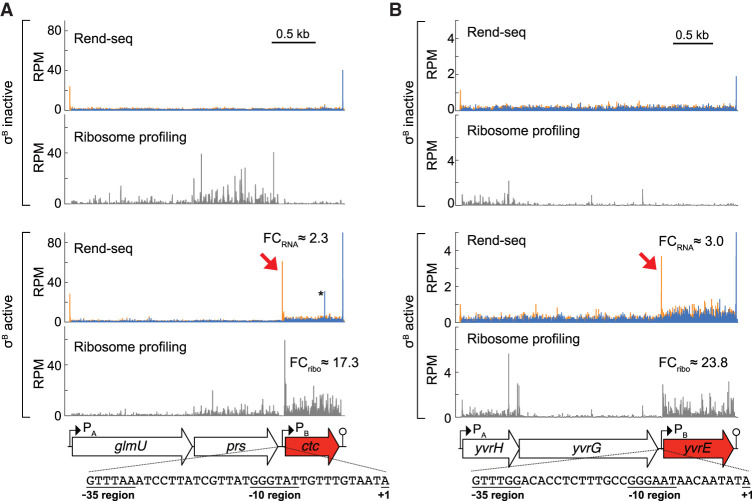
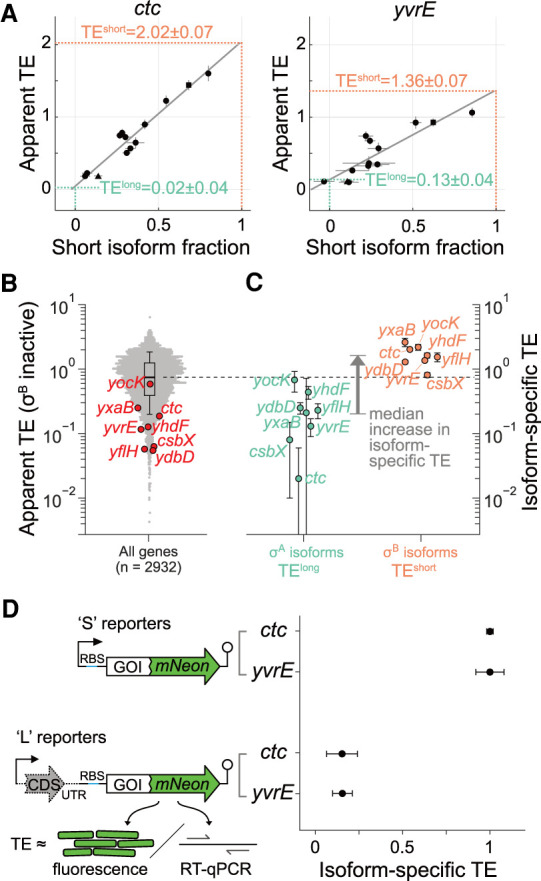
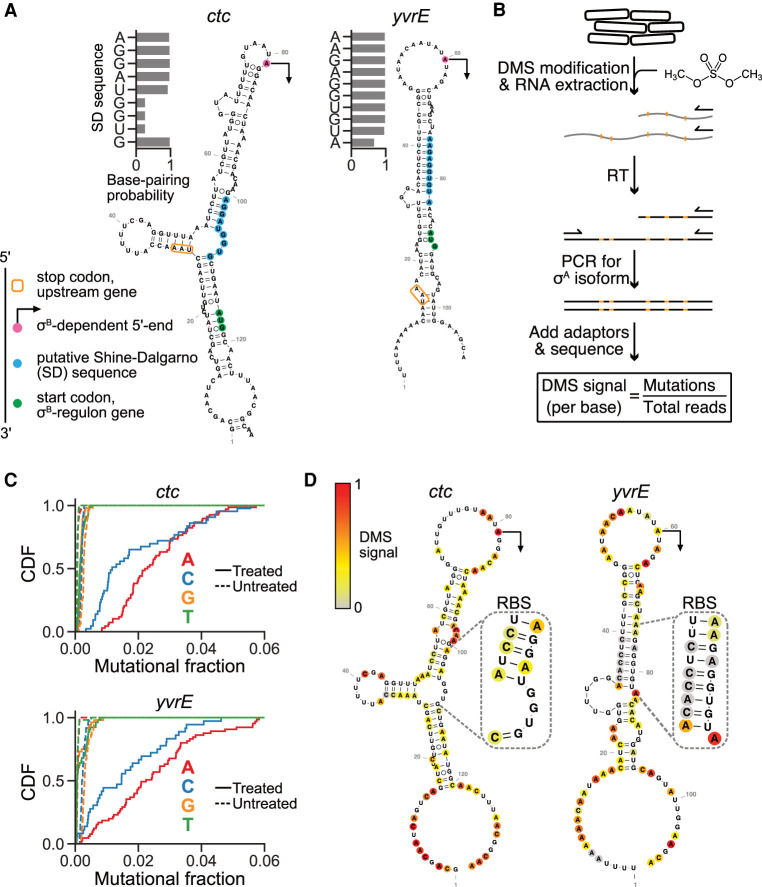
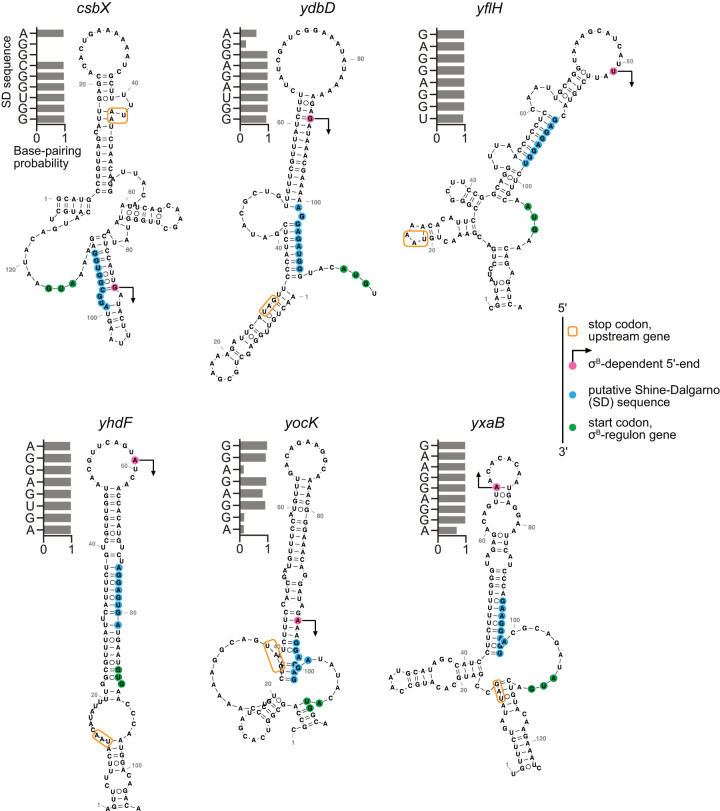
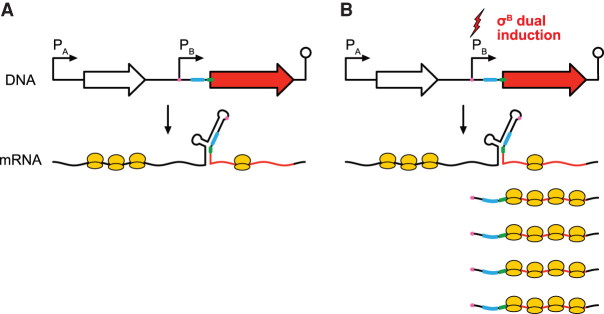
Sigma factors are an important class of bacterial transcription factors that lend specificity to RNA polymerases by binding to distinct promoter elements for genes in their regulons. Here we show that activation of the general stress sigma factor, σB, in Bacillus subtilis paradoxically leads to dramatic induction of translation for a subset of its regulon genes. These genes are translationally repressed when transcribed by the housekeeping sigma factor, σA, owing to extended RNA secondary structures as determined in vivo using DMS-MaPseq. Transcription from σB-dependent promoters ablates the secondary structures and activates translation, leading to dual induction. Translation efficiencies between σB- and σA-dependent RNA isoforms can vary by up to 100-fold, which in multiple cases exceeds the magnitude of transcriptional induction. These results highlight the role of long-range RNA folding in modulating translation and demonstrate that a transcription factor can regulate protein synthesis beyond its effects on transcript levels.
Keywords: B. subtilis; RNA structure; dual induction; sigma factor; translation efficiency.
Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
Similar articles
-
σI from Bacillus subtilis: Impact on Gene Expression and Characterization of σI-Dependent Transcription That Requires New Types of Promoters with Extended -35 and -10 Elements.J Bacteriol. 2018 Aug 10;200(17):e00251-18. doi: 10.1128/JB.00251-18. Print 2018 Sep 1. J Bacteriol. 2018. PMID: 29914988 Free PMC article.
-
Defining the Bacillus subtilis sigma(W) regulon: a comparative analysis of promoter consensus search, run-off transcription/macroarray analysis (ROMA), and transcriptional profiling approaches.J Mol Biol. 2002 Feb 22;316(3):443-57. doi: 10.1006/jmbi.2001.5372. J Mol Biol. 2002. PMID: 11866510
-
Bacillus subtilis sigma 28 and Escherichia coli sigma 32 (htpR) are minor sigma factors that display an overlapping promoter specificity.J Biol Chem. 1985 Feb 25;260(4):2038-41. J Biol Chem. 1985. PMID: 3918995
-
The sigma factors of Bacillus subtilis.Microbiol Rev. 1995 Mar;59(1):1-30. doi: 10.1128/mr.59.1.1-30.1995. Microbiol Rev. 1995. PMID: 7708009 Free PMC article. Review.
-
Heat-shock and general stress response in Bacillus subtilis.Mol Microbiol. 1996 Feb;19(3):417-28. doi: 10.1046/j.1365-2958.1996.396932.x. Mol Microbiol. 1996. PMID: 8830234 Review.
Cited by
-
Spurious regulatory connections dictate the expression-fitness landscape of translation factors.Mol Syst Biol. 2021 Apr;17(4):e10302. doi: 10.15252/msb.202110302. Mol Syst Biol. 2021. PMID: 33900014 Free PMC article.
-
Web-based platform for analysis of RNA folding from high throughput chemical probing data.Nucleic Acids Res. 2022 Jul 5;50(W1):W266-W271. doi: 10.1093/nar/gkac435. Nucleic Acids Res. 2022. PMID: 35657086 Free PMC article.
-
MOB rules: Antibiotic Exposure Reprograms Metabolism to Mobilize Bacillus subtilis in Competitive Interactions.bioRxiv [Preprint]. 2024 Mar 20:2024.03.20.585991. doi: 10.1101/2024.03.20.585991. bioRxiv. 2024. PMID: 38562742 Free PMC article. Preprint.
-
Cross-evaluation of E. coli's operon structures via a whole-cell model suggests alternative cellular benefits for low- versus high-expressing operons.Cell Syst. 2024 Mar 20;15(3):227-245.e7. doi: 10.1016/j.cels.2024.02.002. Epub 2024 Feb 27. Cell Syst. 2024. PMID: 38417437
-
Quantitative Control for Stoichiometric Protein Synthesis.Annu Rev Microbiol. 2021 Oct 8;75:243-267. doi: 10.1146/annurev-micro-041921-012646. Epub 2021 Aug 3. Annu Rev Microbiol. 2021. PMID: 34343023 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases