

Roles of Inflammasomes in Epstein-Barr Virus-Associated Nasopharyngeal Cancer
- PMID: 33918087
- PMCID: PMC8069343
- DOI: 10.3390/cancers13081786
Roles of Inflammasomes in Epstein-Barr Virus-Associated Nasopharyngeal Cancer
Abstract
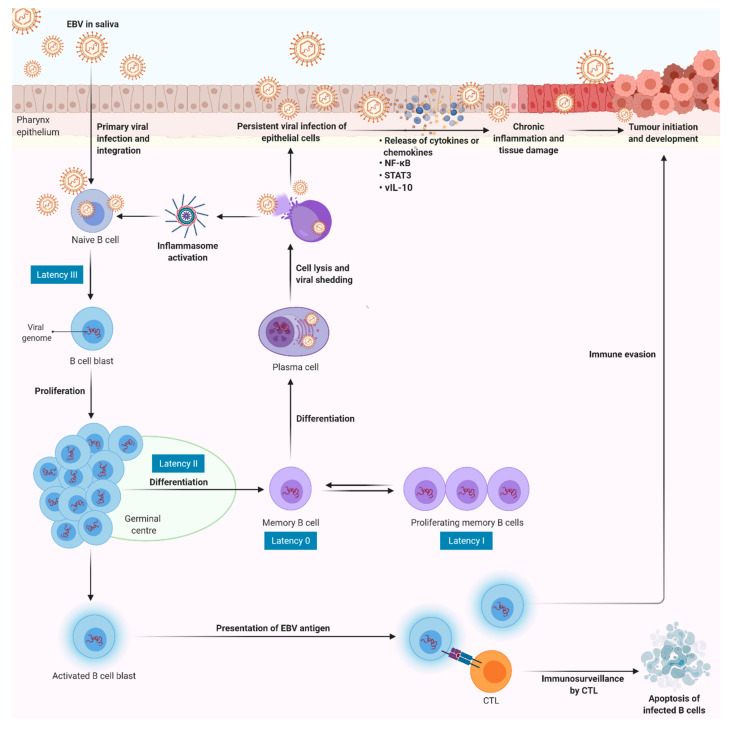
Epstein-Barr virus (EBV) infection is recognised as one of the causative agents in most nasopharyngeal carcinoma (NPC) cases. Expression of EBV viral antigens can induce host's antiviral immune response by activating the inflammasomes to produce pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and IL-18. These cytokines are known to be detrimental to a wide range of virus-infected cells, in which they can activate an inflammatory cell death program, called pyroptosis. However, aberrant inflammasome activation and production of its downstream cytokines lead to chronic inflammation that may contribute to various diseases, including NPC. In this review, we summarise the roles of inflammasomes during viral infection, how EBV evades inflammasome-mediated immune response, and progress into tumourigenesis. The contrasting roles of inflammasomes in cancer, as well as the current therapeutic approaches used in targeting inflammasomes, are also discussed in this review. While the inflammasomes appear to have dual roles in carcinogenesis, there are still many questions that remain unanswered. In particular, the exact molecular mechanism responsible for the regulation of the inflammasomes during carcinogenesis of EBV-associated NPC has not been explored thoroughly. Furthermore, the current practical application of inflammasome inhibitors is limited to specific tumour types, hence, further studies are warranted to discover the potential of targeting the inflammasomes for the treatment of NPC.
Keywords: Epstein–Barr virus; cancer; immune response; inflammasome; inflammation; nasopharyngeal carcinoma; viral evasion.
Conflict of interest statement
The authors declare no conflict of interest. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organisation, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/World Health Organisation.
Figures
Similar articles
-
Tumour inflammasome-derived IL-1β recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma.EMBO Mol Med. 2012 Dec;4(12):1276-93. doi: 10.1002/emmm.201201569. Epub 2012 Oct 15. EMBO Mol Med. 2012. PMID: 23065753 Free PMC article.
-
Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells.J Virol. 2013 Aug;87(15):8606-23. doi: 10.1128/JVI.00805-13. Epub 2013 May 29. J Virol. 2013. PMID: 23720728 Free PMC article.
-
Epstein-Barr virus infection-induced inflammasome activation in human monocytes.PLoS One. 2017 Apr 3;12(4):e0175053. doi: 10.1371/journal.pone.0175053. eCollection 2017. PLoS One. 2017. PMID: 28369146 Free PMC article.
-
Updates on Epstein-Barr Virus (EBV)-Associated Nasopharyngeal Carcinoma: Emphasis on the Latent Gene Products of EBV.Medicina (Kaunas). 2022 Dec 20;59(1):2. doi: 10.3390/medicina59010002. Medicina (Kaunas). 2022. PMID: 36676626 Free PMC article. Review.
-
The role of Epstein-Barr virus infection in the pathogenesis of nasopharyngeal carcinoma.Virol Sin. 2015 Apr;30(2):107-21. doi: 10.1007/s12250-015-3592-5. Epub 2015 Apr 21. Virol Sin. 2015. PMID: 25910483 Free PMC article. Review.
Cited by
-
Deciphering the Role of Epstein-Barr Virus Latent Membrane Protein 1 in Immune Modulation: A Multifaced Signalling Perspective.Viruses. 2024 Apr 4;16(4):564. doi: 10.3390/v16040564. Viruses. 2024. PMID: 38675906 Free PMC article. Review.
-
Targeting the crosstalk of epigenetic modifications and immune evasion in nasopharyngeal cancer.Cell Biol Toxicol. 2023 Dec;39(6):2501-2526. doi: 10.1007/s10565-023-09830-9. Epub 2023 Sep 27. Cell Biol Toxicol. 2023. PMID: 37755585 Review.
-
Macrophage-derived exosomal miRNA-141 triggers endothelial cell pyroptosis by targeting NLRP3 to accelerate sepsis progression.Int J Immunopathol Pharmacol. 2024 Jan-Dec;38:3946320241234736. doi: 10.1177/03946320241234736. Int J Immunopathol Pharmacol. 2024. PMID: 38652556 Free PMC article.
-
Revolutionizing the treatment for nasopharyngeal cancer: the impact, challenges and strategies of stem cell and genetically engineered cell therapies.Front Immunol. 2024 Oct 10;15:1484535. doi: 10.3389/fimmu.2024.1484535. eCollection 2024. Front Immunol. 2024. PMID: 39450176 Free PMC article. Review.
-
The Crucial Role of NLRP3 Inflammasome in Viral Infection-Associated Fibrosing Interstitial Lung Diseases.Int J Mol Sci. 2021 Sep 28;22(19):10447. doi: 10.3390/ijms221910447. Int J Mol Sci. 2021. PMID: 34638790 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
