

The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation
- PMID: 33859667
- PMCID: PMC8042281
- DOI: 10.3389/fgene.2021.639602
The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation
Erratum in
-
Corrigendum: The Role of Histone Lysine Methylation in the Response of Mammalian Cells to Ionizing Radiation.Front Genet. 2022 Apr 13;13:896771. doi: 10.3389/fgene.2022.896771. eCollection 2022. Front Genet. 2022. PMID: 35495134 Free PMC article.
Abstract
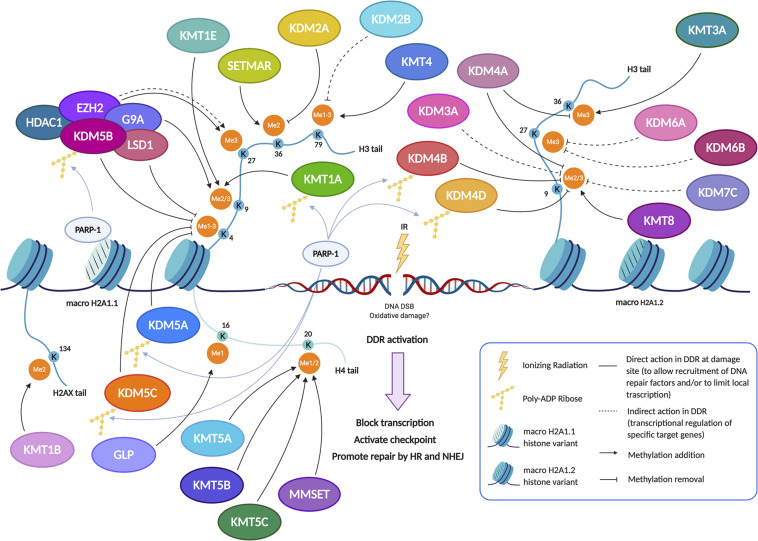
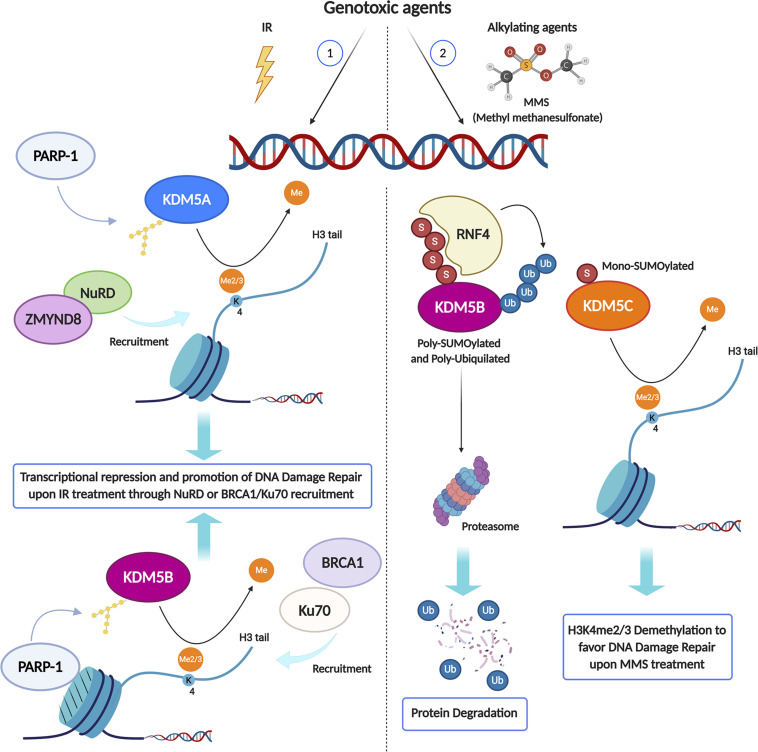
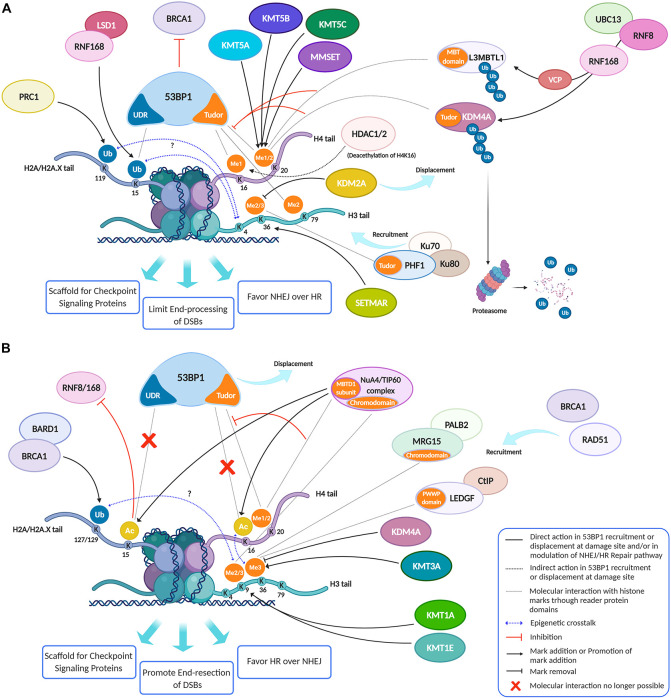
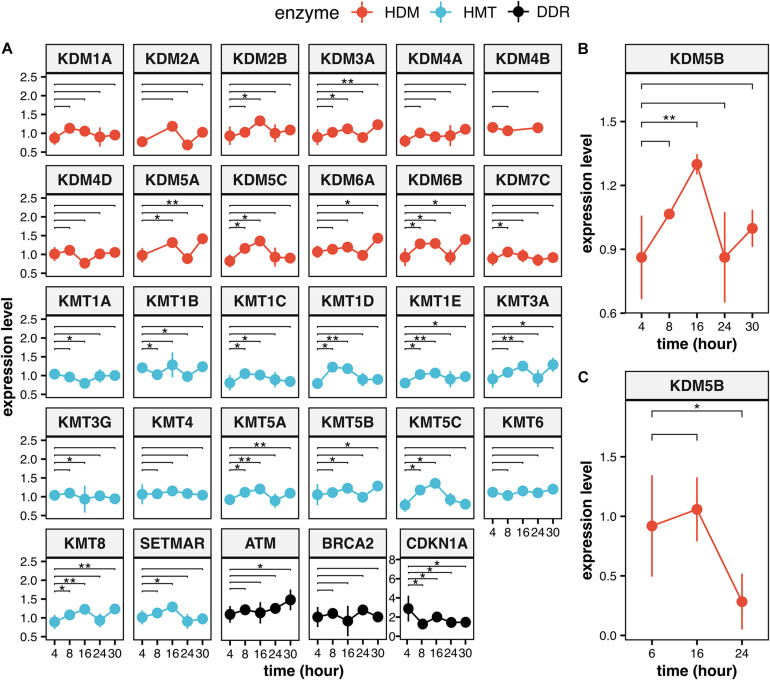
Eukaryotic genomes are wrapped around nucleosomes and organized into different levels of chromatin structure. Chromatin organization has a crucial role in regulating all cellular processes involving DNA-protein interactions, such as DNA transcription, replication, recombination and repair. Histone post-translational modifications (HPTMs) have a prominent role in chromatin regulation, acting as a sophisticated molecular code, which is interpreted by HPTM-specific effectors. Here, we review the role of histone lysine methylation changes in regulating the response to radiation-induced genotoxic damage in mammalian cells. We also discuss the role of histone methyltransferases (HMTs) and histone demethylases (HDMs) and the effects of the modulation of their expression and/or the pharmacological inhibition of their activity on the radio-sensitivity of different cell lines. Finally, we provide a bioinformatic analysis of published datasets showing how the mRNA levels of known HMTs and HDMs are modulated in different cell lines by exposure to different irradiation conditions.
Keywords: DNA damage; DNA repair; HPTMs; histone methylation; ionizing radiation.
Copyright © 2021 Di Nisio, Lupo, Licursi and Negri.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Histone methylation can either promote or reduce cellular radiosensitivity by regulating DNA repair pathways.Mutat Res Rev Mutat Res. 2021 Jan-Jun;787:108362. doi: 10.1016/j.mrrev.2020.108362. Epub 2020 Dec 13. Mutat Res Rev Mutat Res. 2021. PMID: 34083050 Review.
-
Targeting histone methylation and demethylation for non-alcoholic fatty liver disease.Bioorg Chem. 2024 Oct;151:107698. doi: 10.1016/j.bioorg.2024.107698. Epub 2024 Aug 6. Bioorg Chem. 2024. PMID: 39126869 Review.
-
Structure and function of histone methyltransferases.Crit Rev Eukaryot Gene Expr. 2004;14(3):147-69. doi: 10.1615/critreveukaryotgeneexpr.v14.i3.10. Crit Rev Eukaryot Gene Expr. 2004. PMID: 15248813 Review.
-
Histone lysine methylation and chromatin replication.Biochim Biophys Acta. 2014 Dec;1839(12):1433-9. doi: 10.1016/j.bbagrm.2014.03.009. Epub 2014 Mar 28. Biochim Biophys Acta. 2014. PMID: 24686120 Review.
-
Vitamin D and the epigenome.Front Physiol. 2014 Apr 29;5:164. doi: 10.3389/fphys.2014.00164. eCollection 2014. Front Physiol. 2014. PMID: 24808866 Free PMC article. Review.
Cited by
-
To Erase or Not to Erase: Non-Canonical Catalytic Functions and Non-Catalytic Functions of Members of Histone Lysine Demethylase Families.Int J Mol Sci. 2024 Jun 24;25(13):6900. doi: 10.3390/ijms25136900. Int J Mol Sci. 2024. PMID: 39000010 Free PMC article. Review.
-
The Chromatin Landscape around DNA Double-Strand Breaks in Yeast and Its Influence on DNA Repair Pathway Choice.Int J Mol Sci. 2023 Feb 7;24(4):3248. doi: 10.3390/ijms24043248. Int J Mol Sci. 2023. PMID: 36834658 Free PMC article. Review.
-
Unraveling the Complex Interplay between Alpha-Synuclein and Epigenetic Modification.Int J Mol Sci. 2023 Apr 2;24(7):6645. doi: 10.3390/ijms24076645. Int J Mol Sci. 2023. PMID: 37047616 Free PMC article. Review.
-
Epigenetic regulation of the nuclear genome associated with mitochondrial dysfunction in Leber's hereditary optic neuropathy (LHON).Hum Genome Var. 2024 Jan 25;11(1):6. doi: 10.1038/s41439-023-00258-5. Hum Genome Var. 2024. PMID: 38272864 Free PMC article.
-
Stress Responses as Master Keys to Epigenomic Changes in Transcriptome and Metabolome for Cancer Etiology and Therapeutics.Mol Cell Biol. 2022 Jan 20;42(1):e0048321. doi: 10.1128/MCB.00483-21. Epub 2021 Nov 8. Mol Cell Biol. 2022. PMID: 34748401 Free PMC article.
References
-
- Alonso-de Vega I., Paz-Cabrera M. C., Rother M. B., Wiegant W. W., Checa-Rodríguez C., Hernández-Fernaud J. R., et al. (2020). PHF2 regulates homology-directed DNA repair by controlling the resection of DNA double strand breaks. Nucleic Acids Res. 48 4915–4927. 10.1093/nar/gkaa196 - DOI - PMC - PubMed
-
- Amundson S. A., Bittner M., Meltzer P., Trent J., Fornace A. J. (2001). Induction of gene expression as a monitor of exposure to ionizing radiation. Radiat. Res. 156 657–661. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources