

New alignment-based sequence extraction software (ALiBaSeq) and its utility for deep level phylogenetics
- PMID: 33850647
- PMCID: PMC8019319
- DOI: 10.7717/peerj.11019
New alignment-based sequence extraction software (ALiBaSeq) and its utility for deep level phylogenetics
Abstract
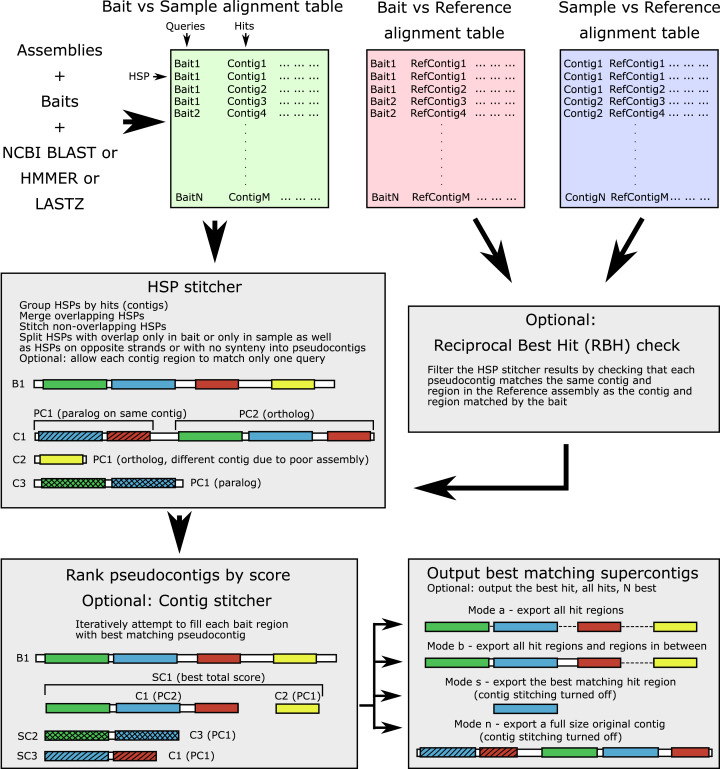
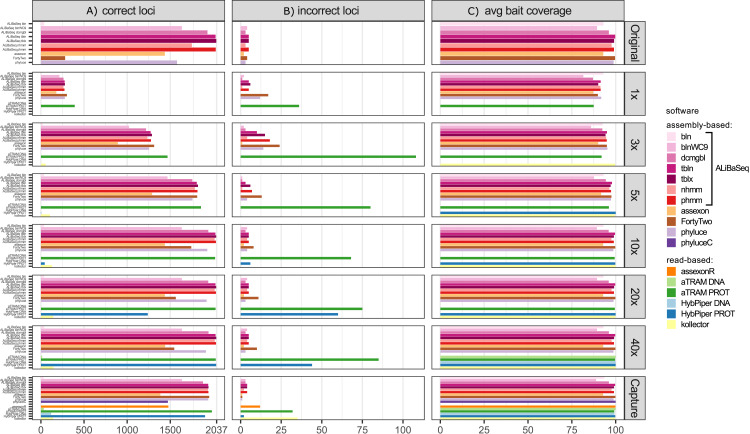
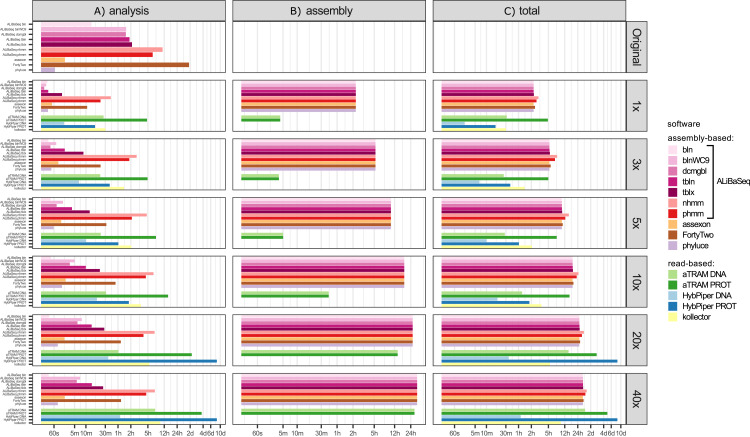
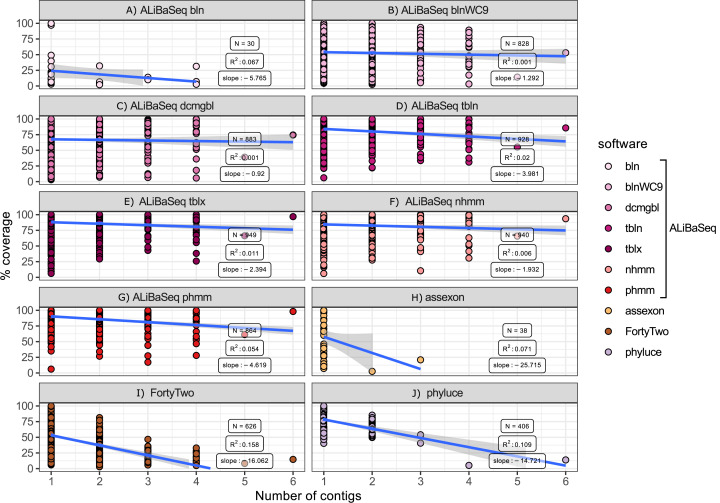
Despite many bioinformatic solutions for analyzing sequencing data, few options exist for targeted sequence retrieval from whole genomic sequencing (WGS) data with the ultimate goal of generating a phylogeny. Available tools especially struggle at deep phylogenetic levels and necessitate amino-acid space searches, which may increase rates of false positive results. Many tools are also difficult to install and may lack adequate user resources. Here, we describe a program that uses freely available similarity search tools to find homologs in assembled WGS data with unparalleled freedom to modify parameters. We evaluate its performance compared to other commonly used bioinformatics tools on two divergent insect species (>200 My) for which annotated genomes exist, and on one large set each of highly conserved and more variable loci. Our software is capable of retrieving orthologs from well-curated or unannotated, low or high depth shotgun, and target capture assemblies as well or better than other software as assessed by recovering the most genes with maximal coverage and with a low rate of false positives throughout all datasets. When assessing this combination of criteria, ALiBaSeq is frequently the best evaluated tool for gathering the most comprehensive and accurate phylogenetic alignments on all types of data tested. The software (implemented in Python), tutorials, and manual are freely available at https://github.com/AlexKnyshov/alibaseq.
Keywords: Alignment; BLAST; HMMER; OrthoDB; Orthology; Phylogenomics; UCE.
© 2021 Knyshov et al.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



Similar articles
-
Patchwork: Alignment-Based Retrieval and Concatenation of Phylogenetic Markers from Genomic Data.Genome Biol Evol. 2023 Dec 1;15(12):evad227. doi: 10.1093/gbe/evad227. Genome Biol Evol. 2023. PMID: 38085033 Free PMC article.
-
MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics.Mol Ecol Resour. 2020 Jul;20(4):892-905. doi: 10.1111/1755-0998.13160. Epub 2020 Apr 25. Mol Ecol Resour. 2020. PMID: 32243090 Free PMC article.
-
morFeus: a web-based program to detect remotely conserved orthologs using symmetrical best hits and orthology network scoring.BMC Bioinformatics. 2014 Aug 6;15(1):263. doi: 10.1186/1471-2105-15-263. BMC Bioinformatics. 2014. PMID: 25096057 Free PMC article.
-
Putting the genome in insect phylogenomics.Curr Opin Insect Sci. 2019 Dec;36:111-117. doi: 10.1016/j.cois.2019.08.002. Epub 2019 Aug 13. Curr Opin Insect Sci. 2019. PMID: 31546095 Review.
-
A guide to bioinformatics for immunologists.Front Immunol. 2013 Dec 4;4:416. doi: 10.3389/fimmu.2013.00416. Front Immunol. 2013. PMID: 24363654 Free PMC article. Review.
Cited by
-
Chromosome-Aware Phylogenomics of Assassin Bugs (Hemiptera: Reduvioidea) Elucidates Ancient Gene Conflict.Mol Biol Evol. 2023 Aug 3;40(8):msad168. doi: 10.1093/molbev/msad168. Mol Biol Evol. 2023. PMID: 37494292 Free PMC article.
-
Genomic Approaches to Uncovering the Coevolutionary History of Parasitic Lice.Life (Basel). 2022 Sep 16;12(9):1442. doi: 10.3390/life12091442. Life (Basel). 2022. PMID: 36143478 Free PMC article. Review.
-
Low hybridization temperatures improve target capture success of invertebrate loci: a case study of leaf-footed bugs (Hemiptera: Coreoidea).R Soc Open Sci. 2023 Jun 28;10(6):230307. doi: 10.1098/rsos.230307. eCollection 2023 Jun. R Soc Open Sci. 2023. PMID: 37388308 Free PMC article.
-
Patchwork: Alignment-Based Retrieval and Concatenation of Phylogenetic Markers from Genomic Data.Genome Biol Evol. 2023 Dec 1;15(12):evad227. doi: 10.1093/gbe/evad227. Genome Biol Evol. 2023. PMID: 38085033 Free PMC article.
-
Mitonuclear compatibility is maintained despite relaxed selection on male mitochondrial DNA in bivalves with doubly uniparental inheritance.Evolution. 2024 Oct 28;78(11):1790-1803. doi: 10.1093/evolut/qpae108. Evolution. 2024. PMID: 38995057
References
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
-
- Barbitoff YA, Polev DE, Glotov AS, Serebryakova EA, Shcherbakova IV, Kiselev AM, Kostareva AA, Glotov OS, Predeus AV. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Scientific Reports. 2020;10(1):418. doi: 10.1038/s41598-020-59026-y. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
