

Axenfeld-Rieger syndrome combined with a foveal anomaly in a three-generation family: a case report
- PMID: 33781219
- PMCID: PMC8008669
- DOI: 10.1186/s12886-021-01899-2
Axenfeld-Rieger syndrome combined with a foveal anomaly in a three-generation family: a case report
Abstract
Background: Axenfeld-Rieger syndrome (ARS) is a rare autosomal dominant eye disorder that can also affect other organs of the human body. The condition is primarily characterized by the anterior segmental abnormalities of the eye. Here, we present an observational case series of a three-generation family with ARS and unexpected foveal anomaly.
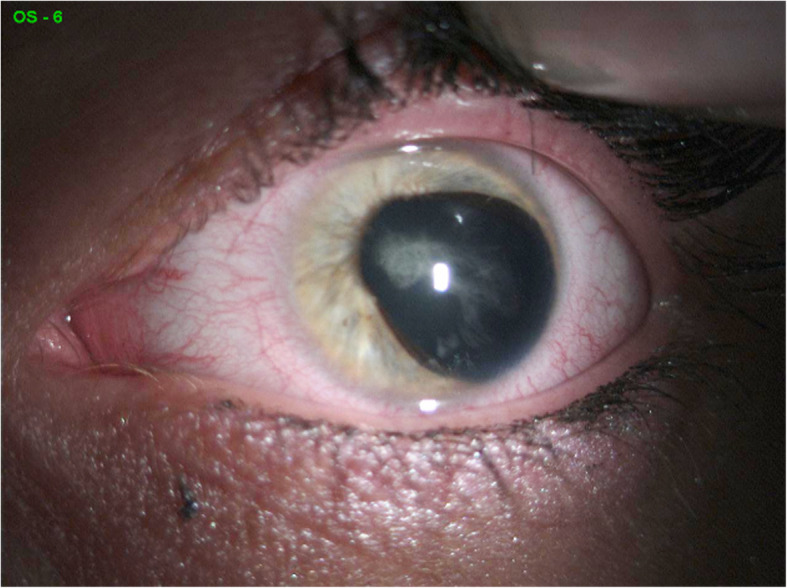
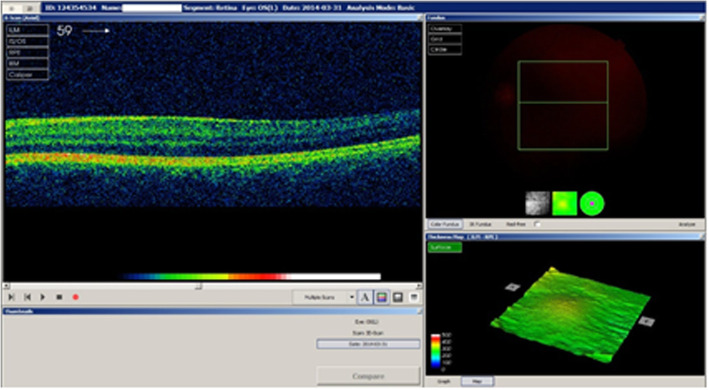
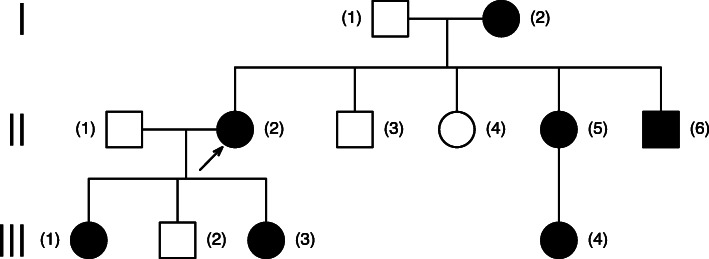
Case presentation: A 33-year-old woman was admitted to an Ophthalmology Clinic in Bialystok for left eye congenital cataract surgery. The patient (proband) was diagnosed with visual deterioration, multiple defects of iris, corectopia, displacement of the Schwalbe's line, and phenotypic characteristics of ARS. A perimetric examination indicated peripheral visual field loss and signs typical for glaucoma. Based on the phenotypic symptoms and genetic test, the patient was diagnosed with Axenfeld Rieger Syndrome. However, the optical coherence tomography of the macula showed foveal anomaly (absence of the physiological pit), which is not typically associated with this genetic disorder. The patient's family history revealed that her two daughters were undergoing treatment for congenital glaucoma, and one of the daughters also had foveal anomaly the same as her mother. Interestingly, an examination of the patient's mother showed typical phenotypic features of ARS such as a defect of the iris, posterior embryotoxon, and coloboma, as well as foveal anomaly. A genetic test confirmed PITX2 mutation in both, proband's two daughters and mother.
Conclusions: This study highlights the occurrence of ARS with unusual ophthalmic features such as foveal anomaly (absence of the physiological pit) in a three-generation family. Although ARS is known to represent the developmental defects of the anterior segment of the eye, it is very important to perform fundus evaluation to identify associated posterior segment anomalies that may affect visual acuity. The presence of ocular defects not typically associated with ARS suggests a wide spectrum of mutations within PITX2 gene which are required to identify in order to determine genotype- phenotype correlation in ARS affected individuals.
Keywords: Axenfeld‐rieger syndrome; Corectopia; Foveal hypoplasia; Hypodontia; Posterior embryotoxon; Rieger anomaly; glaucoma.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
In Vivo Assessment of Retinal Phenotypes in Axenfeld-Rieger Syndrome.Invest Ophthalmol Vis Sci. 2024 Apr 1;65(4):20. doi: 10.1167/iovs.65.4.20. Invest Ophthalmol Vis Sci. 2024. PMID: 38587439 Free PMC article.
-
Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives.Clin Ophthalmol. 2023 Mar 10;17:819-828. doi: 10.2147/OPTH.S379853. eCollection 2023. Clin Ophthalmol. 2023. PMID: 36926528 Free PMC article. Review.
-
Posterior segment findings in Axenfeld-Rieger syndrome.J AAPOS. 2022 Dec;26(6):320-322. doi: 10.1016/j.jaapos.2022.08.263. Epub 2022 Sep 22. J AAPOS. 2022. PMID: 36152758
-
Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation.Mol Vis. 2011;17:1231-8. Epub 2011 May 6. Mol Vis. 2011. PMID: 21617748 Free PMC article.
-
Axenfeld-Rieger syndrome.Clin Genet. 2018 Jun;93(6):1123-1130. doi: 10.1111/cge.13148. Epub 2018 Jan 25. Clin Genet. 2018. PMID: 28972279 Review.
Cited by
-
Diagnostic Challenges of Axenfeld-Rieger Syndrome and a Novel FOXC1 Gene Mutation in a Polish Family.J Clin Med. 2024 Sep 27;13(19):5761. doi: 10.3390/jcm13195761. J Clin Med. 2024. PMID: 39407821 Free PMC article.
-
In Vivo Assessment of Retinal Phenotypes in Axenfeld-Rieger Syndrome.Invest Ophthalmol Vis Sci. 2024 Apr 1;65(4):20. doi: 10.1167/iovs.65.4.20. Invest Ophthalmol Vis Sci. 2024. PMID: 38587439 Free PMC article.
-
Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives.Clin Ophthalmol. 2023 Mar 10;17:819-828. doi: 10.2147/OPTH.S379853. eCollection 2023. Clin Ophthalmol. 2023. PMID: 36926528 Free PMC article. Review.
-
Axenfeld-Rieger syndrome: orthopedic and orthodontic management in a pediatric patient: a case report.Head Face Med. 2022 Jul 8;18(1):25. doi: 10.1186/s13005-022-00329-y. Head Face Med. 2022. PMID: 35804381 Free PMC article.
References
-
- Berenstein-Aizman G, Hazan-Molina H, Drori D, Aizenbud D. Axenfeld-Rieger syndrome: dentofacial manifestation and oral rehabilitation considerations. Pediatr Dent. 2011;33:440–4. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
