

Screening potential FDA-approved inhibitors of the SARS-CoV-2 major protease 3CLpro through high-throughput virtual screening and molecular dynamics simulation
- PMID: 33678621
- PMCID: PMC7993695
- DOI: 10.18632/aging.202703
Screening potential FDA-approved inhibitors of the SARS-CoV-2 major protease 3CLpro through high-throughput virtual screening and molecular dynamics simulation
Abstract
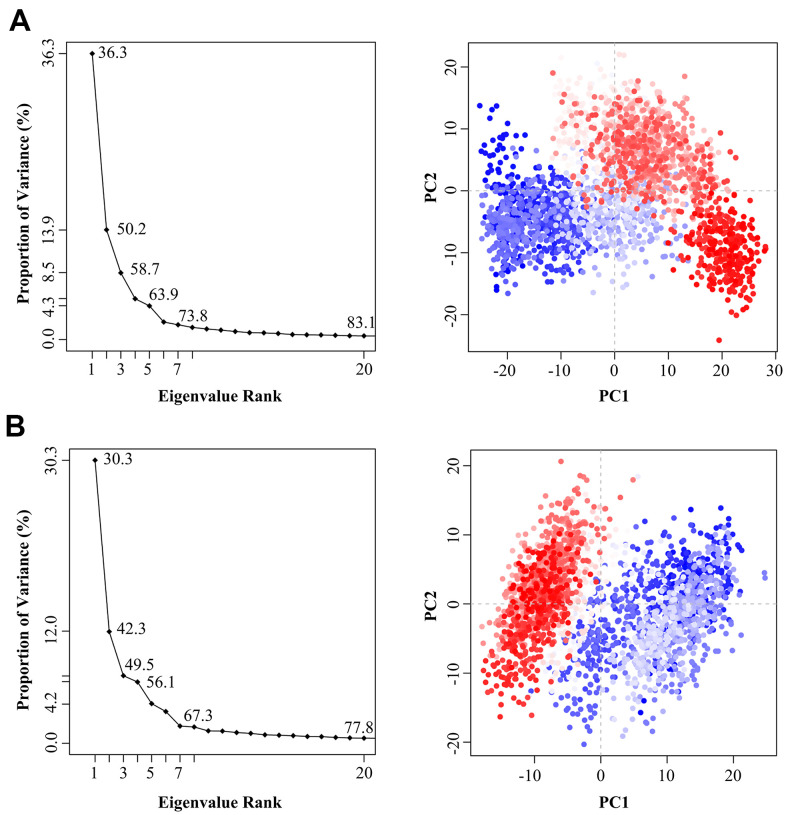
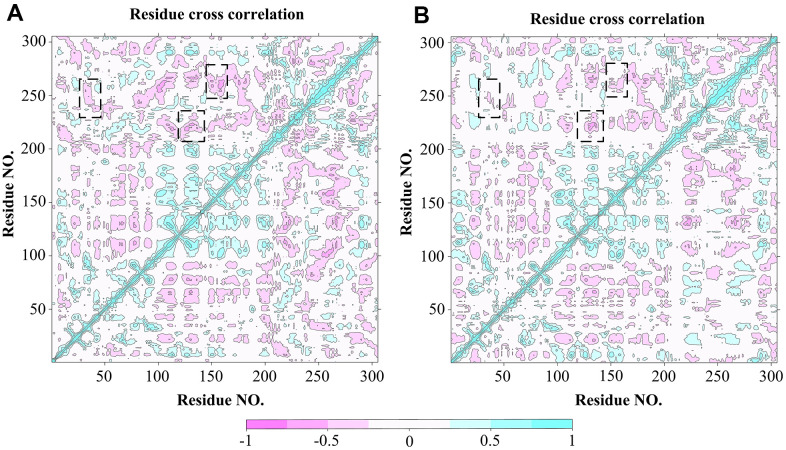
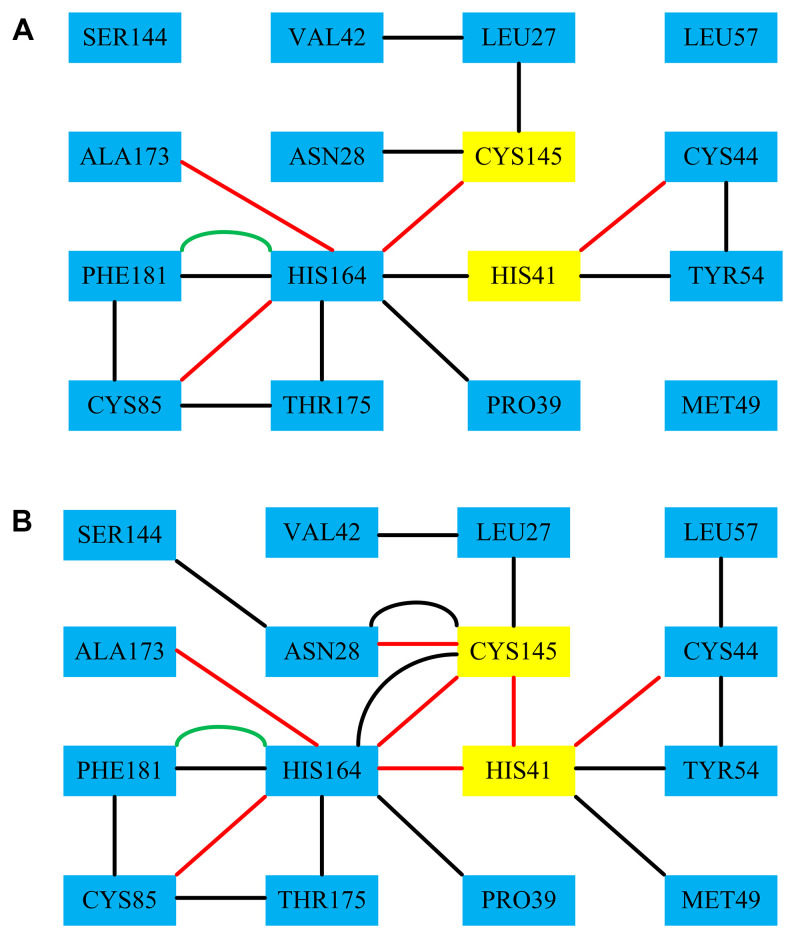
It has been confirmed that the new coronavirus SARS-CoV-2 caused the global pandemic of coronavirus disease 2019 (COVID-19). Studies have found that 3-chymotrypsin-like protease (3CLpro) is an essential enzyme for virus replication, and could be used as a potential target to inhibit SARS-CoV-2. In this work, 3CLpro was used as the target to complete the high-throughput virtual screening of the FDA-approved drugs, and Indinavir and other 10 drugs with high docking scores for 3CLpro were obtained. Studies on the binding pattern of 3CLpro and Indinavir found that Indinavir could form the stable hydrogen bond (H-bond) interactions with the catalytic dyad residues His41-Cys145. Binding free energy study found that Indinavir had high binding affinity with 3CLpro. Subsequently, molecular dynamics simulations were performed on the 3CLpro and 3CLpro-Indinavir systems, respectively. The post-dynamic analyses showed that the conformational state of the 3CLpro-Indinavir system transformed significantly and the system tended to be more stable. Moreover, analyses of the residue interaction network (RIN) and H-bond occupancy revealed that the residue-residue interaction at the catalytic site of 3CLpro was significantly enhanced after binding with Indinavir, which in turn inactivated the protein. In short, through this research, we hope to provide more valuable clues against COVID-19.
Keywords: COVID-19; Indinavir; SARS-CoV-2; molecular docking; molecular dynamics simulation.
Conflict of interest statement
Figures
Similar articles
-
Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations.Int J Mol Sci. 2021 Aug 24;22(17):9124. doi: 10.3390/ijms22179124. Int J Mol Sci. 2021. PMID: 34502033 Free PMC article.
-
Discovery of New Hydroxyethylamine Analogs against 3CLpro Protein Target of SARS-CoV-2: Molecular Docking, Molecular Dynamics Simulation, and Structure-Activity Relationship Studies.J Chem Inf Model. 2020 Dec 28;60(12):5754-5770. doi: 10.1021/acs.jcim.0c00326. Epub 2020 Jun 18. J Chem Inf Model. 2020. PMID: 32551639
-
Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases.Eur J Pharmacol. 2021 Jun 15;901:174082. doi: 10.1016/j.ejphar.2021.174082. Epub 2021 Apr 3. Eur J Pharmacol. 2021. PMID: 33823185 Free PMC article.
-
An Updated Review on SARS-CoV-2 Main Proteinase (MPro): Protein Structure and Small-Molecule Inhibitors.Curr Top Med Chem. 2021;21(6):442-460. doi: 10.2174/1568026620666201207095117. Curr Top Med Chem. 2021. PMID: 33292134 Review.
-
SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors.Biomolecules. 2021 Apr 19;11(4):607. doi: 10.3390/biom11040607. Biomolecules. 2021. PMID: 33921886 Free PMC article. Review.
Cited by
-
Pharmacophore Oriented MP2 Characterization of Charge Distribution for Anti-SARS-CoV-2 Inhibitor Nirmatrelvir.J Mol Struct. 2023 Oct 15;1290:135871. doi: 10.1016/j.molstruc.2023.135871. Epub 2023 May 30. J Mol Struct. 2023. PMID: 37313328 Free PMC article.
-
Recent Advances in Nitrogen-Containing Heterocyclic Scaffolds as Antiviral Agents.Med Chem. 2024;20(5):487-502. doi: 10.2174/0115734064280150231212113012. Med Chem. 2024. PMID: 38279757 Review.
-
Studying Conformational Properties of Transmembrane Domain of KCNE3 in a Lipid Bilayer Membrane Using Molecular Dynamics Simulations.Membranes (Basel). 2024 Feb 4;14(2):45. doi: 10.3390/membranes14020045. Membranes (Basel). 2024. PMID: 38392672 Free PMC article.
-
Identification of potential target endoribonuclease NSP15 inhibitors of SARS-COV-2 from natural products through high-throughput virtual screening and molecular dynamics simulation.J Food Biochem. 2022 May;46(5):e14085. doi: 10.1111/jfbc.14085. Epub 2022 Feb 6. J Food Biochem. 2022. PMID: 35128681 Free PMC article.
-
Receptor-Based Pharmacophore Modelling of a series of ligands used as inhibitors of the SARS-CoV-2 virus by complementary theoretical approaches, molecular docking, and reactivity descriptors.F1000Res. 2023 Jun 26;12:749. doi: 10.12688/f1000research.133426.1. eCollection 2023. F1000Res. 2023. PMID: 39291142 Free PMC article.
References
-
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, et al., and China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020; 382:727–33. 10.1056/NEJMoa2001017 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
