

Dominant mutations in MIEF1 affect mitochondrial dynamics and cause a singular late onset optic neuropathy
- PMID: 33632269
- PMCID: PMC7905578
- DOI: 10.1186/s13024-021-00431-w
Dominant mutations in MIEF1 affect mitochondrial dynamics and cause a singular late onset optic neuropathy
Abstract
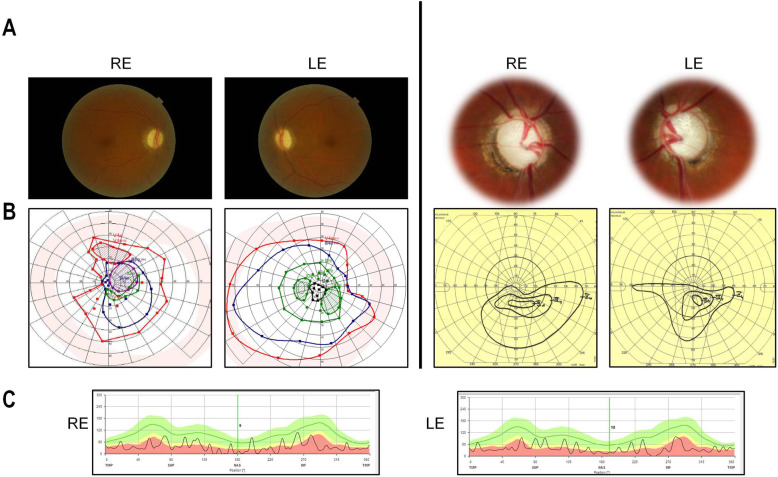
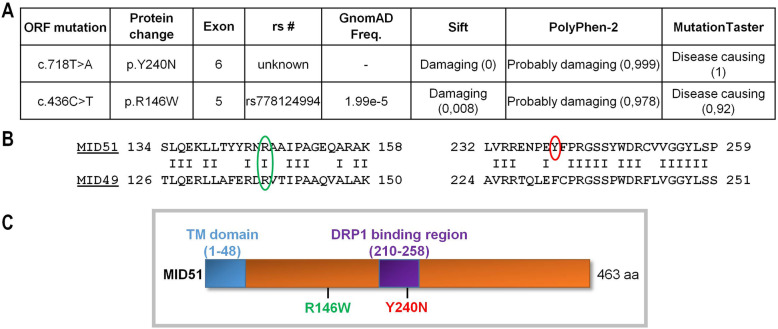
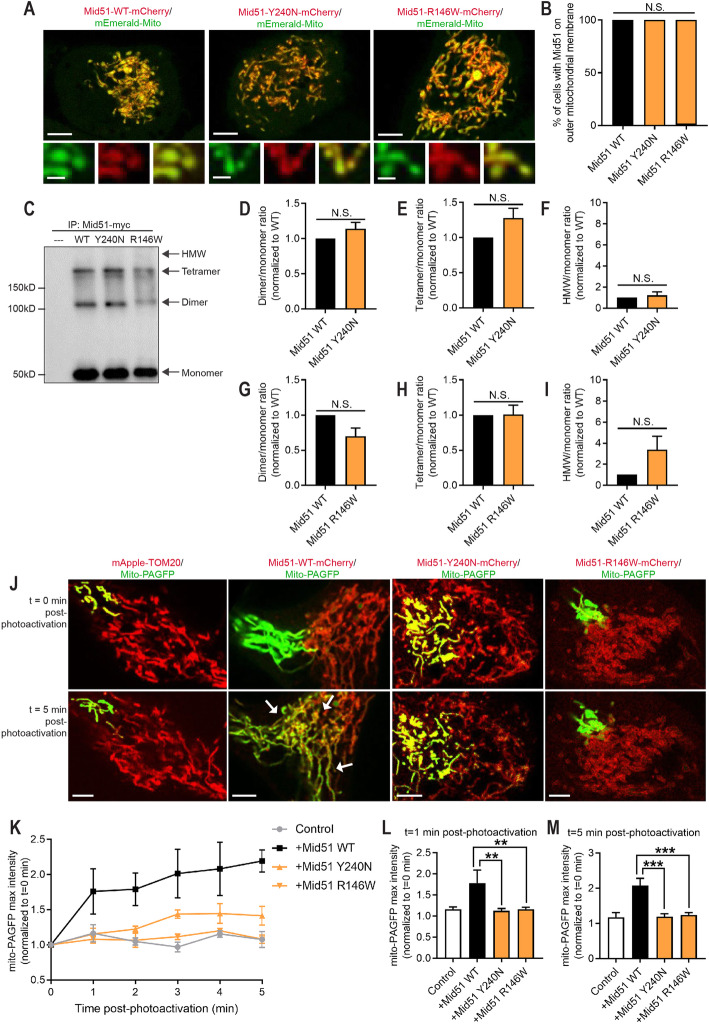
Inherited optic neuropathies are the most common mitochondrial diseases, leading to neurodegeneration involving the irreversible loss of retinal ganglion cells, optic nerve degeneration and central visual loss. Importantly, properly regulated mitochondrial dynamics are critical for maintaining cellular homeostasis, and are further regulated by MIEF1 (mitochondrial elongation factor 1) which encodes for MID51 (mitochondrial dynamics protein 51), an outer mitochondrial membrane protein that acts as an adaptor protein to regulate mitochondrial fission. However, dominant mutations in MIEF1 have not been previously linked to any human disease. Using targeted sequencing of genes involved in mitochondrial dynamics, we report the first heterozygous variants in MIEF1 linked to disease, which cause an unusual form of late-onset progressive optic neuropathy characterized by the initial loss of peripheral visual fields. Pathogenic MIEF1 variants linked to optic neuropathy do not disrupt MID51's localization to the outer mitochondrial membrane or its oligomerization, but rather, significantly disrupt mitochondrial network dynamics compared to wild-type MID51 in high spatial and temporal resolution confocal microscopy live imaging studies. Together, our study identifies dominant MIEF1 mutations as a cause for optic neuropathy and further highlights the important role of properly regulated mitochondrial dynamics in neurodegeneration.
Keywords: Dominant optic atrophy (DOA); Inherited optic neuropathy (ION); MIEF1; Mid51; Mitochondria dynamics; Mitochondrial disease; Neurodegeneration; Peripheral visual field.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy.Autophagy. 2019 Dec;15(12):2107-2125. doi: 10.1080/15548627.2019.1596494. Epub 2019 Mar 28. Autophagy. 2019. PMID: 30894073 Free PMC article.
-
The mitochondrial elongation factors MIEF1 and MIEF2 exert partially distinct functions in mitochondrial dynamics.Exp Cell Res. 2013 Nov 1;319(18):2893-904. doi: 10.1016/j.yexcr.2013.07.010. Epub 2013 Jul 20. Exp Cell Res. 2013. PMID: 23880462
-
Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission.EMBO J. 2011 Jun 24;30(14):2762-78. doi: 10.1038/emboj.2011.198. EMBO J. 2011. PMID: 21701560 Free PMC article.
-
Mitochondrial Membrane Dynamics and Inherited Optic Neuropathies.In Vivo. 2017 Jul-Aug;31(4):511-525. doi: 10.21873/invivo.11090. In Vivo. 2017. PMID: 28652416 Free PMC article. Review.
-
The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease.Clin Sci (Lond). 2016 Nov 1;130(21):1861-74. doi: 10.1042/CS20160030. Clin Sci (Lond). 2016. PMID: 27660309 Review.
Cited by
-
Genetic spectrum and characteristics of autosomal optic neuropathy in Korean: Use of next-generation sequencing in suspected hereditary optic atrophy.Front Neurol. 2022 Aug 22;13:978532. doi: 10.3389/fneur.2022.978532. eCollection 2022. Front Neurol. 2022. PMID: 36071901 Free PMC article.
-
The Role of Mitochondria in Optic Atrophy With Autosomal Inheritance.Front Neurosci. 2021 Nov 15;15:784987. doi: 10.3389/fnins.2021.784987. eCollection 2021. Front Neurosci. 2021. PMID: 34867178 Free PMC article. Review.
-
Molecular Mechanisms behind Inherited Neurodegeneration of the Optic Nerve.Biomolecules. 2021 Mar 25;11(4):496. doi: 10.3390/biom11040496. Biomolecules. 2021. PMID: 33806088 Free PMC article. Review.
-
Players in Mitochondrial Dynamics and Female Reproduction.Front Mol Biosci. 2021 Oct 11;8:717328. doi: 10.3389/fmolb.2021.717328. eCollection 2021. Front Mol Biosci. 2021. PMID: 34708072 Free PMC article. Review.
-
Super-resolution microscopy: Insights into mitochondria-lysosome crosstalk in health and disease.J Cell Biol. 2023 Dec 4;222(12):e202305032. doi: 10.1083/jcb.202305032. Epub 2023 Nov 2. J Cell Biol. 2023. PMID: 37917024 Free PMC article.
References
-
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
