

What is the Pathogenic CAG Expansion Length in Huntington's Disease?
- PMID: 33579866
- PMCID: PMC7990448
- DOI: 10.3233/JHD-200445
What is the Pathogenic CAG Expansion Length in Huntington's Disease?
Abstract
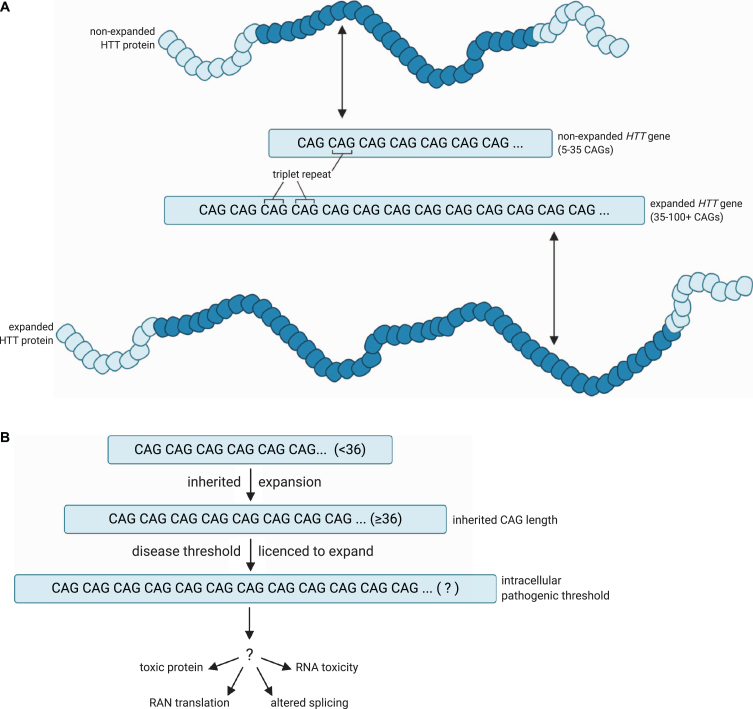
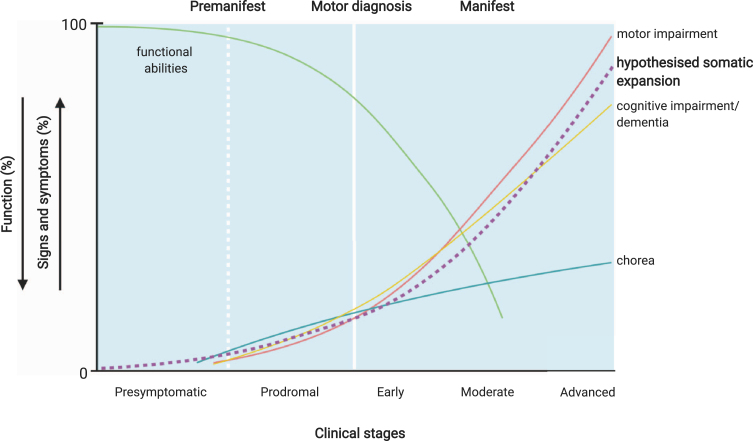
Huntington's disease (HD) (OMIM 143100) is caused by an expanded CAG repeat tract in the HTT gene. The inherited CAG length is known to expand further in somatic and germline cells in HD subjects. Age at onset of the disease is inversely correlated with the inherited CAG length, but is further modulated by a series of genetic modifiers which are most likely to act on the CAG repeat in HTT that permit it to further expand. Longer repeats are more prone to expansions, and this expansion is age dependent and tissue-specific. Given that the inherited tract expands through life and most subjects develop disease in mid-life, this implies that in cells that degenerate, the CAG length is likely to be longer than the inherited length. These findings suggest two thresholds- the inherited CAG length which permits further expansion, and the intracellular pathogenic threshold, above which cells become dysfunctional and die. This two-step mechanism has been previously proposed and modelled mathematically to give an intracellular pathogenic threshold at a tract length of 115 CAG (95% confidence intervals 70- 165 CAG). Empirically, the intracellular pathogenic threshold is difficult to determine. Clues from studies of people and models of HD, and from other diseases caused by expanded repeat tracts, place this threshold between 60- 100 CAG, most likely towards the upper part of that range. We assess this evidence and discuss how the intracellular pathogenic threshold in manifest disease might be better determined. Knowing the cellular pathogenic threshold would be informative for both understanding the mechanism in HD and deploying treatments.
Keywords: DNA repair; DNA repeat expansion; Huntington’s disease; spinocerebellar ataxias; trinucleotide repeat.
Conflict of interest statement
LJ is a member of the Scientific Advisory Boards of LoQus23 Therapeutics and Triplet Therapeutics. JD, SP, NR and PH have no conflicts of interest.
Figures
Similar articles
-
A CAG repeat threshold for therapeutics targeting somatic instability in Huntington's disease.Brain. 2024 May 3;147(5):1784-1798. doi: 10.1093/brain/awae063. Brain. 2024. PMID: 38387080 Free PMC article.
-
Patterns of CAG repeat instability in the central nervous system and periphery in Huntington's disease and in spinocerebellar ataxia type 1.Hum Mol Genet. 2020 Aug 29;29(15):2551-2567. doi: 10.1093/hmg/ddaa139. Hum Mol Genet. 2020. PMID: 32761094 Free PMC article.
-
DNA repair in the trinucleotide repeat disorders.Lancet Neurol. 2017 Jan;16(1):88-96. doi: 10.1016/S1474-4422(16)30350-7. Lancet Neurol. 2017. PMID: 27979358 Review.
-
Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington's disease knock-in mice is blocked by Mlh1 knock-out.Hum Mol Genet. 2020 Nov 4;29(18):3044-3053. doi: 10.1093/hmg/ddaa196. Hum Mol Genet. 2020. PMID: 32876667 Free PMC article.
-
Methods for Assessing DNA Repair and Repeat Expansion in Huntington's Disease.Methods Mol Biol. 2018;1780:483-495. doi: 10.1007/978-1-4939-7825-0_22. Methods Mol Biol. 2018. PMID: 29856032 Review.
Cited by
-
Modifier pathways in polyglutamine (PolyQ) diseases: from genetic screens to drug targets.Cell Mol Life Sci. 2022 May 3;79(5):274. doi: 10.1007/s00018-022-04280-8. Cell Mol Life Sci. 2022. PMID: 35503478 Free PMC article. Review.
-
Genetic modifiers of repeat expansion disorders.Emerg Top Life Sci. 2023 Dec 14;7(3):325-337. doi: 10.1042/ETLS20230015. Emerg Top Life Sci. 2023. PMID: 37861103 Free PMC article.
-
Huntingtin is an RNA binding protein and participates in NEAT1-mediated paraspeckles.Sci Adv. 2024 Jul 19;10(29):eado5264. doi: 10.1126/sciadv.ado5264. Epub 2024 Jul 19. Sci Adv. 2024. PMID: 39028820 Free PMC article.
-
Mutant huntingtin confers cell-autonomous phenotypes on Huntington's disease iPSC-derived microglia.Sci Rep. 2023 Nov 22;13(1):20477. doi: 10.1038/s41598-023-46852-z. Sci Rep. 2023. PMID: 37993517 Free PMC article.
-
The flavonoid luteolin reduces mutant huntingtin aggregation and cytotoxicity in huntingtin-mutated neuroblastoma cells.Saudi Pharm J. 2023 Dec;31(12):101871. doi: 10.1016/j.jsps.2023.101871. Epub 2023 Nov 21. Saudi Pharm J. 2023. PMID: 38125952 Free PMC article.
References
-
- Hannan AJ. Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet. 2018;19(5):286–98. - PubMed
-
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Prim. 2015;1(1):15005. - PubMed
-
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30(1):575–621. - PubMed
-
- Coutelier M, Coarelli G, Monin ML, Konop J, Davoine CS, Tesson C, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelo-pathies. Brain. 2017;140(6):1579–94. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
