

Non-Transferrin-Bound Iron in the Spotlight: Novel Mechanistic Insights into the Vasculotoxic and Atherosclerotic Effect of Iron
- PMID: 33554718
- PMCID: PMC8328045
- DOI: 10.1089/ars.2020.8167
Non-Transferrin-Bound Iron in the Spotlight: Novel Mechanistic Insights into the Vasculotoxic and Atherosclerotic Effect of Iron
Abstract
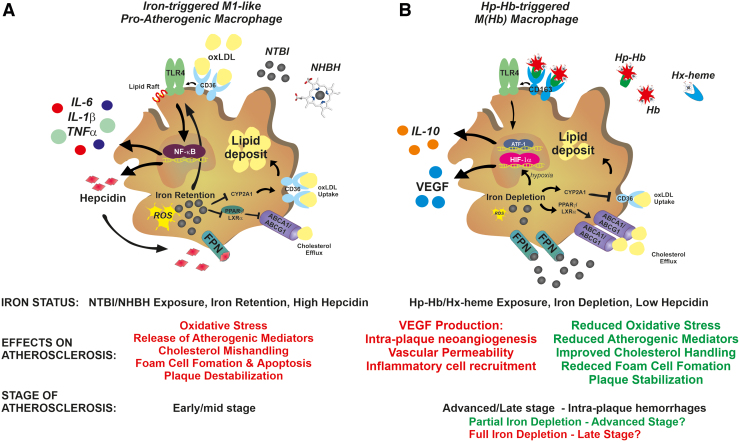
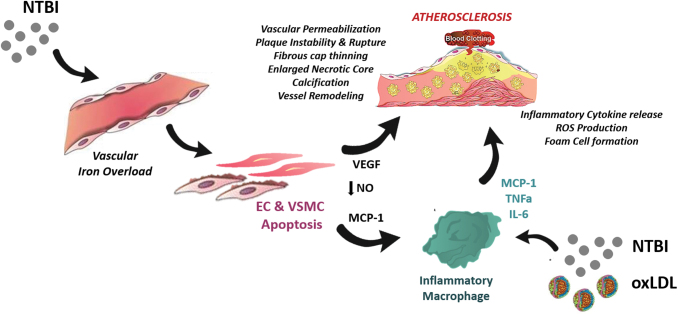
Significance: While atherosclerosis is an almost inevitable consequence of aging, food preferences, lack of exercise, and other aspects of the lifestyle in many countries, the identification of new risk factors is of increasing importance to tackle a disease, which has become a major health burden for billions of people. Iron has long been suspected to promote the development of atherosclerosis, but data have been conflicting, and the contribution of iron is still debated controversially. Recent Advances: Several experimental and clinical studies have been recently published about this longstanding controversial problem, highlighting the critical need to unravel the complexity behind this topic. Critical Issues: The aim of the current review is to provide an overview of the current knowledge about the proatherosclerotic impact of iron, and discuss the emerging role of non-transferrin-bound iron (NTBI) as driver of vasculotoxicity and atherosclerosis. Finally, I will provide detailed mechanistic insights on the cellular processes and molecular pathways underlying iron-exacerbated atherosclerosis. Overall, this review highlights a complex framework where NTBI acts at multiple levels in atherosclerosis by altering the serum and vascular microenvironment in a proatherogenic and proinflammatory manner, affecting the functionality and survival of vascular cells, promoting foam cell formation and inducing angiogenesis, calcification, and plaque destabilization. Future Directions: The use of additional iron markers (e.g., NTBI) may help adequately predict predisposition to cardiovascular disease. Clinical studies are needed in the aging population to address the atherogenic role of iron fluctuations within physiological limits and the therapeutic value of iron restriction approaches. Antioxid. Redox Signal. 35, 387-414.
Keywords: atherosclerosis; calcification; cardiovascular disease; endothelial dysfunction; inflammation; intraplaque macrophages; iron; iron-aggravated atherosclerosis; iron-loaded VSMC; nontransferrin-bound iron (NTBI); oxidized LDL; reactive oxygen species (ROS).
Conflict of interest statement
No competing financial interests exist.
Figures
Similar articles
-
Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction.Eur Heart J. 2020 Jul 21;41(28):2681-2695. doi: 10.1093/eurheartj/ehz112. Eur Heart J. 2020. PMID: 30903157
-
The Role of Iron in Atherosclerosis and its Association with Related Diseases.Curr Atheroscler Rep. 2024 Nov 9;27(1):1. doi: 10.1007/s11883-024-01251-1. Curr Atheroscler Rep. 2024. PMID: 39520606 Review.
-
Iron and Atherosclerosis: The Link Revisited.Trends Mol Med. 2019 Aug;25(8):659-661. doi: 10.1016/j.molmed.2019.05.012. Epub 2019 Jun 20. Trends Mol Med. 2019. PMID: 31230908 Review.
-
Non-transferrin-bound iron is associated with plasma level of soluble intercellular adhesion molecule-1 but not with in vivo low-density lipoprotein oxidation.Atherosclerosis. 2007 Sep;194(1):272-8. doi: 10.1016/j.atherosclerosis.2006.08.012. Epub 2006 Sep 11. Atherosclerosis. 2007. PMID: 16963052
-
Can iron chelators influence the progression of atherosclerosis?Hemoglobin. 2008;32(1-2):123-34. doi: 10.1080/03630260701726871. Hemoglobin. 2008. PMID: 18274990 Review.
Cited by
-
Iron overload and chelation modulates bisretinoid levels in the retina.Front Ophthalmol (Lausanne). 2023 Dec 22;3:1305864. doi: 10.3389/fopht.2023.1305864. eCollection 2023. Front Ophthalmol (Lausanne). 2023. PMID: 38983013 Free PMC article.
-
The Role of Iron in Atherosclerosis in Apolipoprotein E Deficient Mice.Front Cardiovasc Med. 2022 May 20;9:857933. doi: 10.3389/fcvm.2022.857933. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35669479 Free PMC article.
-
Macrophage metabolic rewiring improves heme-suppressed efferocytosis and tissue damage in sickle cell disease.Blood. 2023 Jun 22;141(25):3091-3108. doi: 10.1182/blood.2022018026. Blood. 2023. PMID: 36952641 Free PMC article.
-
Prognostic Factors and Clinical Considerations for Iron Chelation Therapy in Myelodysplastic Syndrome Patients.J Blood Med. 2021 Dec 3;12:1019-1030. doi: 10.2147/JBM.S287876. eCollection 2021. J Blood Med. 2021. PMID: 34887690 Free PMC article. Review.
-
Serum ferritin and the risk of myocardial infarction: A Mendelian randomization study.Medicine (Baltimore). 2024 Apr 26;103(17):e37952. doi: 10.1097/MD.0000000000037952. Medicine (Baltimore). 2024. PMID: 38669402 Free PMC article.
References
-
- Adams PC, Pankow JS, Barton JC, Acton RT, Leiendecker-Foster C, McLaren GD, Speechley M, and Eckfeldt JH. HFE C282Y homozygosity is associated with lower total and low-density lipoprotein cholesterol: the hemochromatosis and iron overload screening study. Circ Cardiovasc Genet 2: 34–37, 2009 - PubMed
-
- Aessopos A, Farmakis D, Tsironi M, Diamanti-Kandarakis E, Matzourani M, Fragodimiri C, Hatziliami A, and Karagiorga M. Endothelial function and arterial stiffness in sickle-thalassemia patients. Atherosclerosis 191: 427–432, 2007 - PubMed
-
- Agarwal A, Balla J, Balla G, Croatt AJ, Vercellotti GM, and Nath KA. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am J Physiol 271: F814–F823, 1996 - PubMed
-
- Ali F, Hamdulay SS, Kinderlerer AR, Boyle JJ, Lidington EA, Yamaguchi T, Soares MP, Haskard DO, Randi AM, and Mason JC. Statin-mediated cytoprotection of human vascular endothelial cells: a role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost 5: 2537–2546, 2007 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
