

Evolutionary dynamics and structural consequences of de novo beneficial mutations and mutant lineages arising in a constant environment
- PMID: 33541358
- PMCID: PMC7863352
- DOI: 10.1186/s12915-021-00954-0
Evolutionary dynamics and structural consequences of de novo beneficial mutations and mutant lineages arising in a constant environment
Abstract
Background: Microbial evolution experiments can be used to study the tempo and dynamics of evolutionary change in asexual populations, founded from single clones and growing into large populations with multiple clonal lineages. High-throughput sequencing can be used to catalog de novo mutations as potential targets of selection, determine in which lineages they arise, and track the fates of those lineages. Here, we describe a long-term experimental evolution study to identify targets of selection and to determine when, where, and how often those targets are hit.
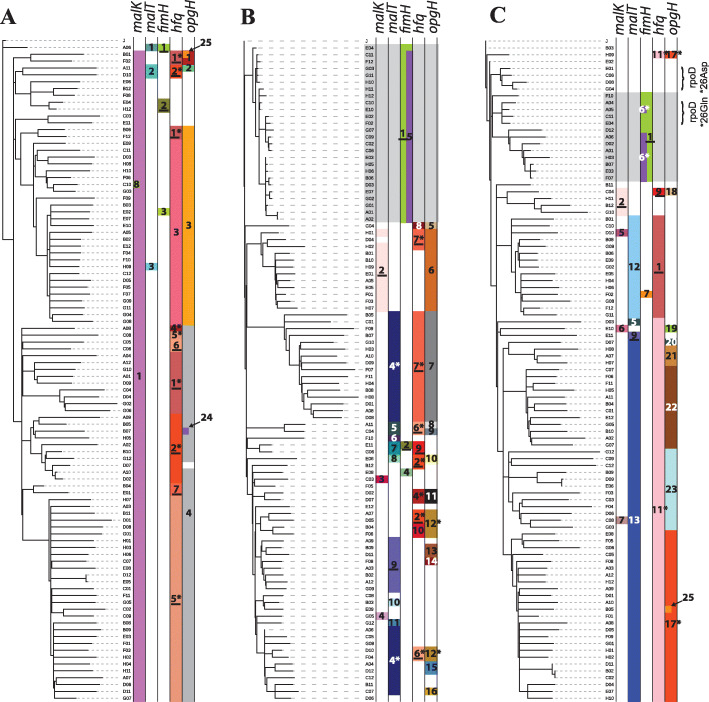
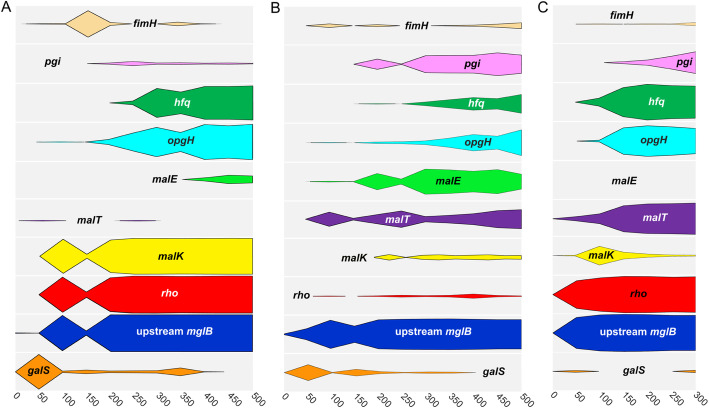
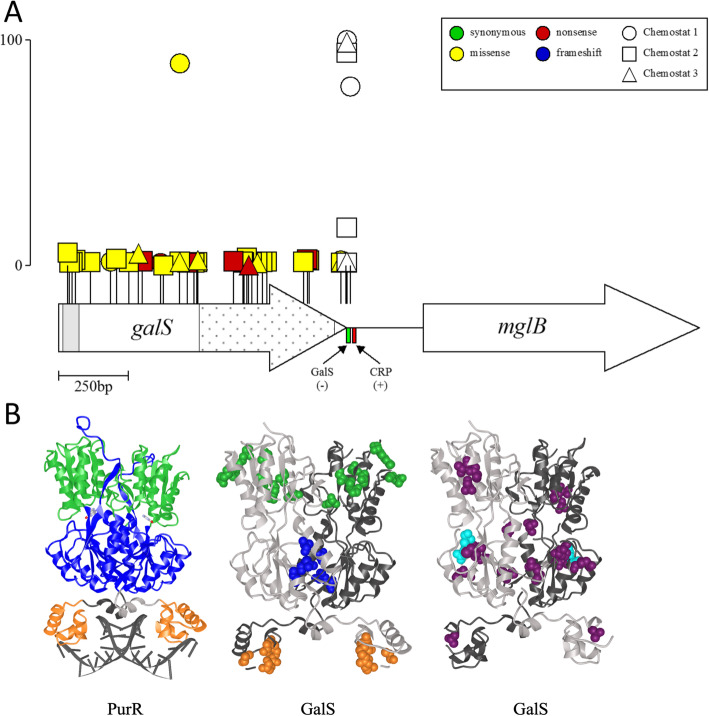
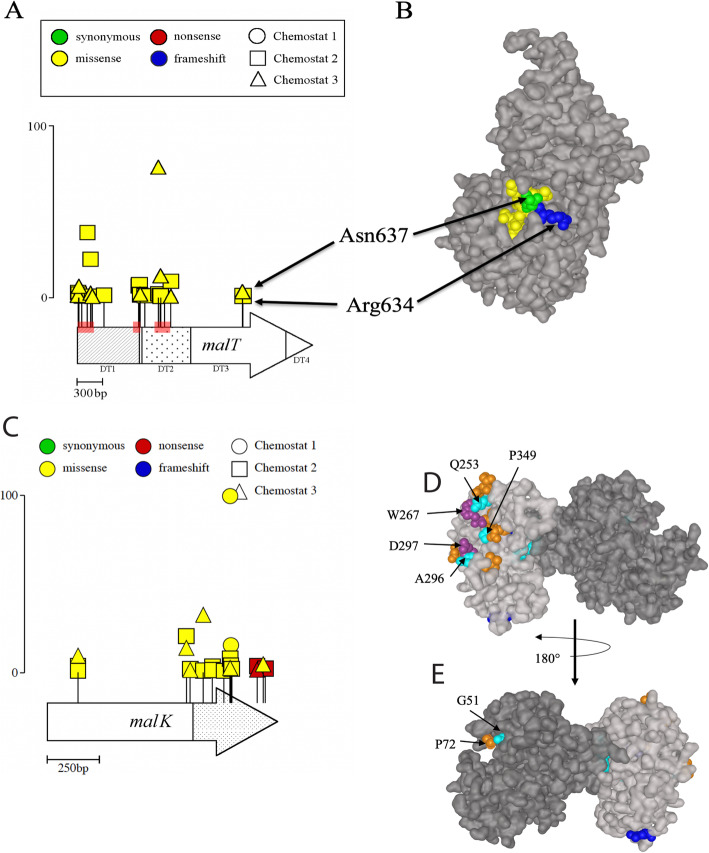
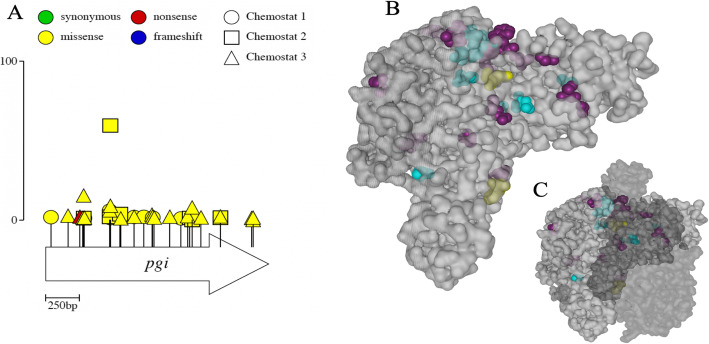
Results: We experimentally evolved replicate Escherichia coli populations that originated from a mutator/nonsense suppressor ancestor under glucose limitation for between 300 and 500 generations. Whole-genome, whole-population sequencing enabled us to catalog 3346 de novo mutations that reached > 1% frequency. We sequenced the genomes of 96 clones from each population when allelic diversity was greatest in order to establish whether mutations were in the same or different lineages and to depict lineage dynamics. Operon-specific mutations that enhance glucose uptake were the first to rise to high frequency, followed by global regulatory mutations. Mutations related to energy conservation, membrane biogenesis, and mitigating the impact of nonsense mutations, both ancestral and derived, arose later. New alleles were confined to relatively few loci, with many instances of identical mutations arising independently in multiple lineages, among and within replicate populations. However, most never exceeded 10% in frequency and were at a lower frequency at the end of the experiment than at their maxima, indicating clonal interference. Many alleles mapped to key structures within the proteins that they mutated, providing insight into their functional consequences.
Conclusions: Overall, we find that when mutational input is increased by an ancestral defect in DNA repair, the spectrum of high-frequency beneficial mutations in a simple, constant resource-limited environment is narrow, resulting in extreme parallelism where many adaptive mutations arise but few ever go to fixation.
Keywords: Adaptation; Clonal interference; E. coli; Experimental evolution; Parallelism; Resource limitation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



Similar articles
-
Tempo and mode of genome evolution in a 50,000-generation experiment.Nature. 2016 Aug 11;536(7615):165-70. doi: 10.1038/nature18959. Epub 2016 Aug 1. Nature. 2016. PMID: 27479321 Free PMC article.
-
Clonal interference and the periodic selection of new beneficial mutations in Escherichia coli.Genetics. 2006 Apr;172(4):2093-100. doi: 10.1534/genetics.105.052373. Epub 2006 Feb 19. Genetics. 2006. PMID: 16489229 Free PMC article.
-
Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations.Genetics. 2002 Oct;162(2):557-66. doi: 10.1093/genetics/162.2.557. Genetics. 2002. PMID: 12399371 Free PMC article.
-
Evolution of global regulatory networks during a long-term experiment with Escherichia coli.Bioessays. 2007 Sep;29(9):846-60. doi: 10.1002/bies.20629. Bioessays. 2007. PMID: 17691099 Review.
-
Genomic investigations of evolutionary dynamics and epistasis in microbial evolution experiments.Curr Opin Genet Dev. 2015 Dec;35:33-9. doi: 10.1016/j.gde.2015.08.008. Epub 2015 Sep 14. Curr Opin Genet Dev. 2015. PMID: 26370471 Free PMC article. Review.
Cited by
-
Uncovering the GacS-mediated role in evolutionary progression through trajectory reconstruction in Pseudomonas aeruginosa.Nucleic Acids Res. 2024 Apr 24;52(7):3856-3869. doi: 10.1093/nar/gkae187. Nucleic Acids Res. 2024. PMID: 38477346 Free PMC article.
-
Patterns of Fitness and Gene Expression Epistasis Generated by Beneficial Mutations in the rho and rpoB Genes of Escherichia coli during High-Temperature Adaptation.Mol Biol Evol. 2024 Sep 4;41(9):msae187. doi: 10.1093/molbev/msae187. Mol Biol Evol. 2024. PMID: 39235107 Free PMC article.
-
Discovery of positive and purifying selection in metagenomic time series of hypermutator microbial populations.PLoS Genet. 2022 Aug 18;18(8):e1010324. doi: 10.1371/journal.pgen.1010324. eCollection 2022 Aug. PLoS Genet. 2022. PMID: 35981004 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
