

Ebola virus triggers receptor tyrosine kinase-dependent signaling to promote the delivery of viral particles to entry-conducive intracellular compartments
- PMID: 33513206
- PMCID: PMC7875390
- DOI: 10.1371/journal.ppat.1009275
Ebola virus triggers receptor tyrosine kinase-dependent signaling to promote the delivery of viral particles to entry-conducive intracellular compartments
Abstract
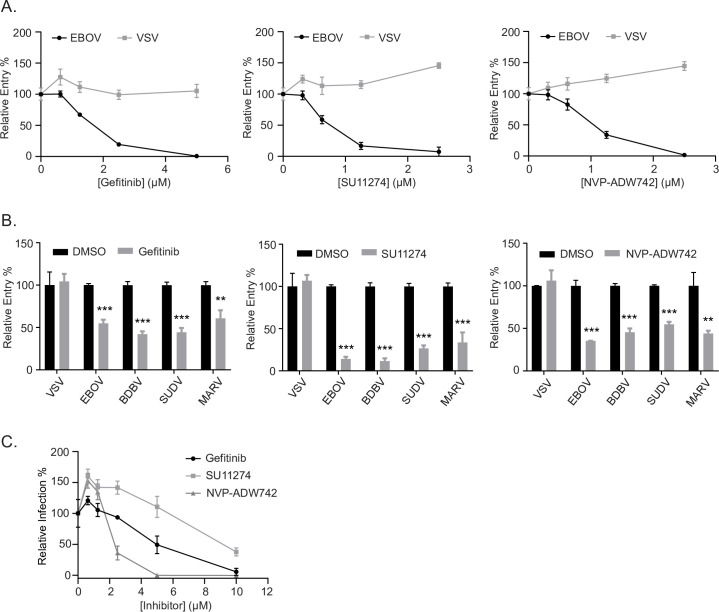
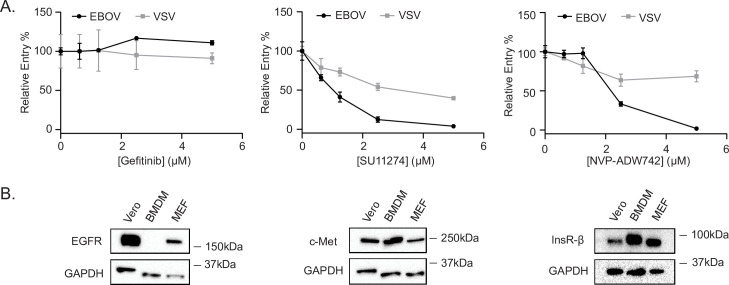
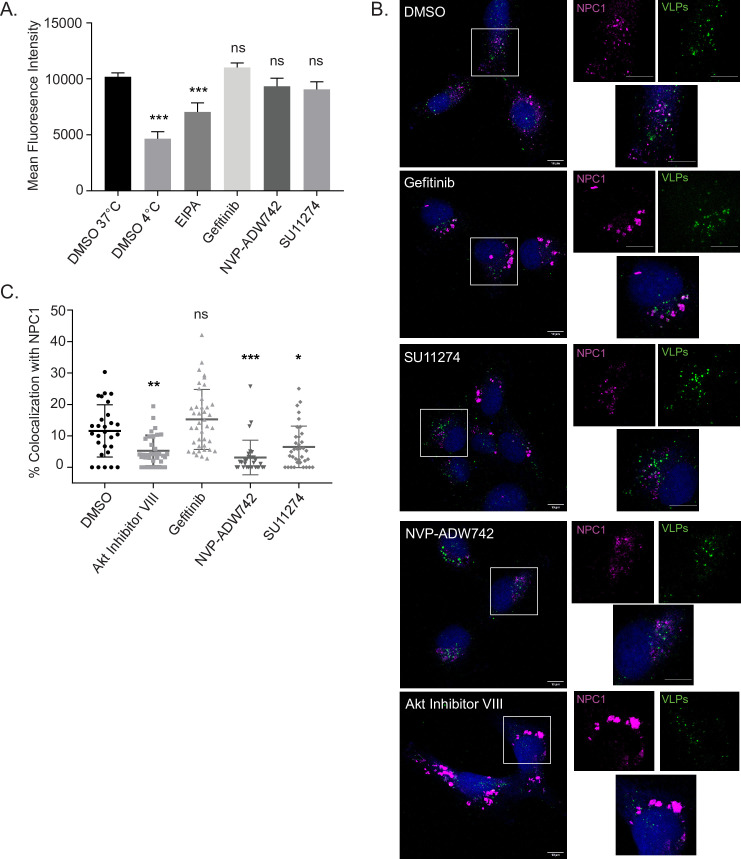
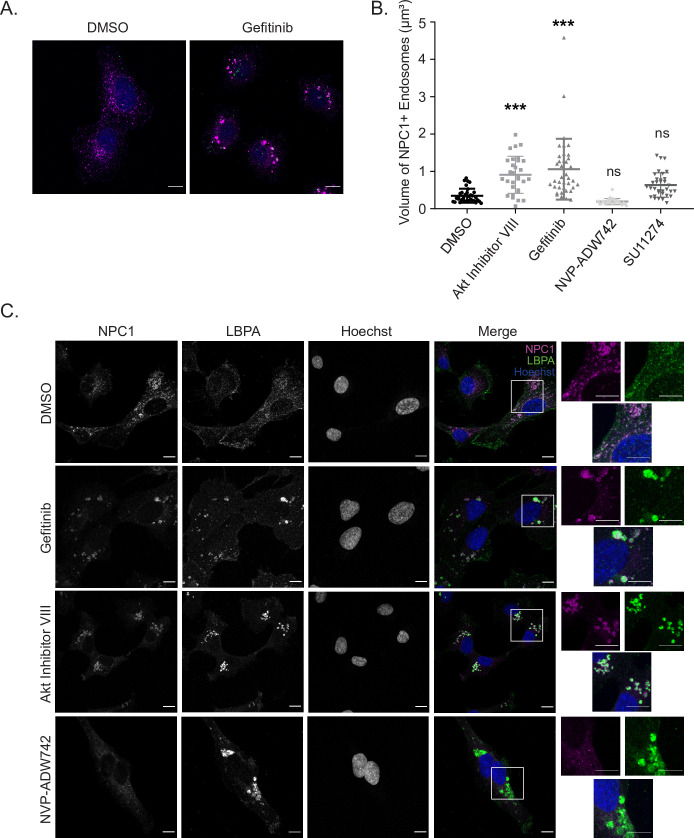
Filoviruses, such as the Ebola virus (EBOV) and Marburg virus (MARV), are causative agents of sporadic outbreaks of hemorrhagic fevers in humans. To infect cells, filoviruses are internalized via macropinocytosis and traffic through the endosomal pathway where host cathepsin-dependent cleavage of the viral glycoproteins occurs. Subsequently, the cleaved viral glycoprotein interacts with the late endosome/lysosome resident host protein, Niemann-Pick C1 (NPC1). This interaction is hypothesized to trigger viral and host membrane fusion, which results in the delivery of the viral genome into the cytoplasm and subsequent initiation of replication. Some studies suggest that EBOV viral particles activate signaling cascades and host-trafficking factors to promote their localization with host factors that are essential for entry. However, the mechanism through which these activating signals are initiated remains unknown. By screening a kinase inhibitor library, we found that receptor tyrosine kinase inhibitors potently block EBOV and MARV GP-dependent viral entry. Inhibitors of epidermal growth factor receptor (EGFR), tyrosine protein kinase Met (c-Met), and the insulin receptor (InsR)/insulin like growth factor 1 receptor (IGF1R) blocked filoviral GP-mediated entry and prevented growth of replicative EBOV in Vero cells. Furthermore, inhibitors of c-Met and InsR/IGF1R also blocked viral entry in macrophages, the primary targets of EBOV infection. Interestingly, while the c-Met and InsR/IGF1R inhibitors interfered with EBOV trafficking to NPC1, virus delivery to the receptor was not impaired in the presence of the EGFR inhibitor. Instead, we observed that the NPC1 positive compartments were phenotypically altered and rendered incompetent to permit viral entry. Despite their different mechanisms of action, all three RTK inhibitors tested inhibited virus-induced Akt activation, providing a possible explanation for how EBOV may activate signaling pathways during entry. In sum, these studies strongly suggest that receptor tyrosine kinases initiate signaling cascades essential for efficient post-internalization entry steps.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



Similar articles
-
Direct Intracellular Visualization of Ebola Virus-Receptor Interaction by In Situ Proximity Ligation.mBio. 2021 Jan 12;12(1):e03100-20. doi: 10.1128/mBio.03100-20. mBio. 2021. PMID: 33436438 Free PMC article.
-
Filoviruses Use the HOPS Complex and UVRAG To Traffic to Niemann-Pick C1 Compartments during Viral Entry.J Virol. 2020 Jul 30;94(16):e01002-20. doi: 10.1128/JVI.01002-20. Print 2020 Jul 30. J Virol. 2020. PMID: 32493822 Free PMC article.
-
Ebola virus and severe acute respiratory syndrome coronavirus display late cell entry kinetics: evidence that transport to NPC1+ endolysosomes is a rate-defining step.J Virol. 2015 Mar;89(5):2931-43. doi: 10.1128/JVI.03398-14. Epub 2014 Dec 31. J Virol. 2015. PMID: 25552710 Free PMC article.
-
Filovirus entry: a novelty in the viral fusion world.Viruses. 2012 Feb;4(2):258-75. doi: 10.3390/v4020258. Epub 2012 Feb 7. Viruses. 2012. PMID: 22470835 Free PMC article. Review.
-
Potential pharmacological strategies targeting the Niemann-Pick C1 receptor and Ebola virus glycoprotein interaction.Eur J Med Chem. 2021 Nov 5;223:113654. doi: 10.1016/j.ejmech.2021.113654. Epub 2021 Jun 19. Eur J Med Chem. 2021. PMID: 34175537 Review.
Cited by
-
Cell-impermeable staurosporine analog targets extracellular kinases to inhibit HSV and SARS-CoV-2.Commun Biol. 2022 Oct 16;5(1):1096. doi: 10.1038/s42003-022-04067-4. Commun Biol. 2022. PMID: 36245045 Free PMC article.
-
Akt Plays Differential Roles during the Life Cycles of Acute and Persistent Murine Norovirus Strains in Macrophages.J Virol. 2022 Feb 9;96(3):e0192321. doi: 10.1128/JVI.01923-21. Epub 2021 Nov 17. J Virol. 2022. PMID: 34787460 Free PMC article.
-
Pseudotyped Viruses for Marburgvirus and Ebolavirus.Adv Exp Med Biol. 2023;1407:105-132. doi: 10.1007/978-981-99-0113-5_6. Adv Exp Med Biol. 2023. PMID: 36920694
-
Groundnut Bud Necrosis Virus Modulates the Expression of Innate Immune, Endocytosis, and Cuticle Development-Associated Genes to Circulate and Propagate in Its Vector, Thrips palmi.Front Microbiol. 2022 Mar 17;13:773238. doi: 10.3389/fmicb.2022.773238. eCollection 2022. Front Microbiol. 2022. PMID: 35369489 Free PMC article.
-
Interferon-Induced HERC5 Inhibits Ebola Virus Particle Production and Is Antagonized by Ebola Glycoprotein.Cells. 2021 Sep 13;10(9):2399. doi: 10.3390/cells10092399. Cells. 2021. PMID: 34572049 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
