

The Kaumoebavirus LCC10 Genome Reveals a Unique Gene Strand Bias among "Extended Asfarviridae"
- PMID: 33498382
- PMCID: PMC7909422
- DOI: 10.3390/v13020148
The Kaumoebavirus LCC10 Genome Reveals a Unique Gene Strand Bias among "Extended Asfarviridae"
Abstract
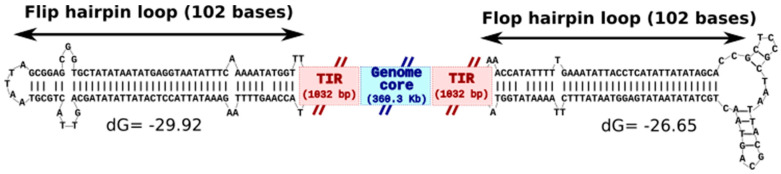
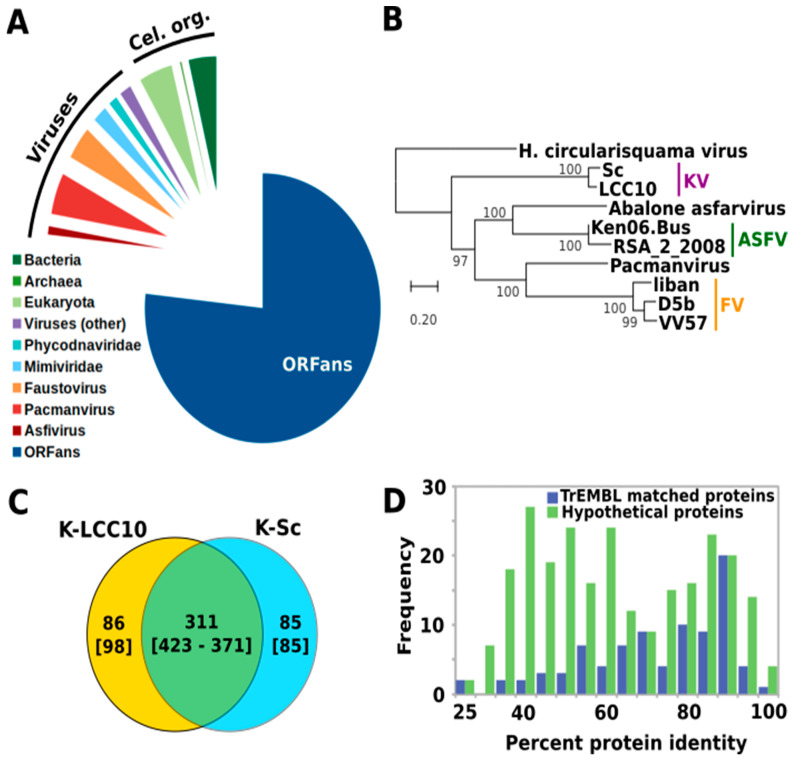
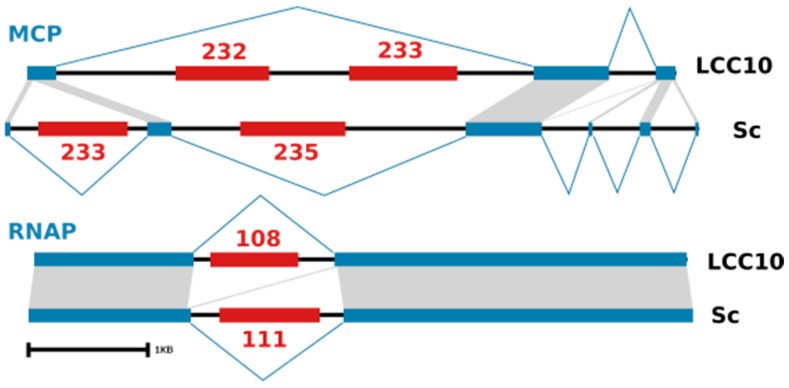
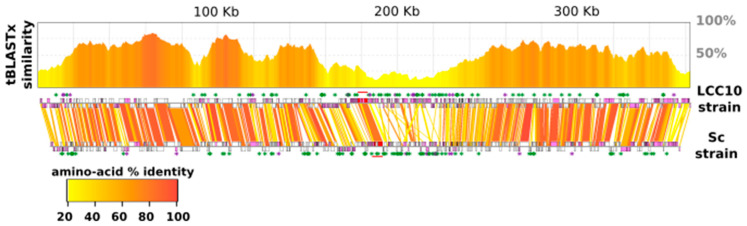
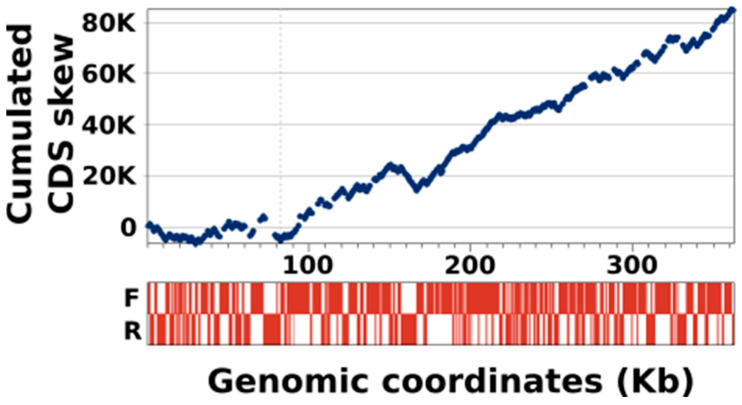
Kaumoebavirus infects the amoeba Vermamoeba vermiformis and has recently been described as a distant relative of the African swine fever virus. To characterize the diversity and evolution of this novel viral genus, we report here on the isolation and genome sequencing of a second strain of Kaumoebavirus, namely LCC10. Detailed analysis of the sequencing data suggested that its 362-Kb genome is linear with covalently closed hairpin termini, so that DNA forms a single continuous polynucleotide chain. Comparative genomic analysis indicated that although the two sequenced Kaumoebavirus strains share extensive gene collinearity, 180 predicted genes were either gained or lost in only one genome. As already observed in another distant relative, i.e., Faustovirus, which infects the same host, the center and extremities of the Kaumoebavirus genome exhibited a higher rate of sequence divergence and the major capsid protein gene was colonized by type-I introns. A possible role of the Vermamoeba host in the genesis of these evolutionary traits is hypothesized. The Kaumoebavirus genome exhibited a significant gene strand bias over the two-third of genome length, a feature not seen in the other members of the "extended Asfarviridae" clade. We suggest that this gene strand bias was induced by a putative single origin of DNA replication located near the genome extremity that imparted a selective force favoring the genes positioned on the leading strand.
Keywords: Kaumoebavirus; Nucleocytoviricota; Vermamoeba vermiformis; extended Asfarviridae; gene strand bias; nucleo-cytoplasmic large DNA virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Asfarviruses and Closely Related Giant Viruses.Viruses. 2023 Apr 20;15(4):1015. doi: 10.3390/v15041015. Viruses. 2023. PMID: 37112995 Free PMC article. Review.
-
Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae.Viruses. 2016 Oct 28;8(11):278. doi: 10.3390/v8110278. Viruses. 2016. PMID: 27801826 Free PMC article.
-
Comparative Genomics Unveils Regionalized Evolution of the Faustovirus Genomes.Viruses. 2020 May 24;12(5):577. doi: 10.3390/v12050577. Viruses. 2020. PMID: 32456325 Free PMC article.
-
Pacmanvirus, a New Giant Icosahedral Virus at the Crossroads between Asfarviridae and Faustoviruses.J Virol. 2017 Jun 26;91(14):e00212-17. doi: 10.1128/JVI.00212-17. Print 2017 Jul 15. J Virol. 2017. PMID: 28446673 Free PMC article.
-
The morphogenesis of different giant viruses as additional evidence for a common origin of Nucleocytoviricota.Curr Opin Virol. 2021 Aug;49:102-110. doi: 10.1016/j.coviro.2021.05.004. Epub 2021 Jun 8. Curr Opin Virol. 2021. PMID: 34116391 Review.
Cited by
-
Diversity of Giant Viruses Infecting Vermamoeba vermiformis.Front Microbiol. 2022 Apr 22;13:808499. doi: 10.3389/fmicb.2022.808499. eCollection 2022. Front Microbiol. 2022. PMID: 35602053 Free PMC article. Review.
-
Morphological and Genomic Features of the New Klosneuvirinae Isolate Fadolivirus IHUMI-VV54.Front Microbiol. 2021 Sep 21;12:719703. doi: 10.3389/fmicb.2021.719703. eCollection 2021. Front Microbiol. 2021. PMID: 34621250 Free PMC article.
-
Analysis of the Genomic Features and Evolutionary History of Pithovirus-Like Isolates Reveals Two Major Divergent Groups of Viruses.J Virol. 2023 Jul 27;97(7):e0041123. doi: 10.1128/jvi.00411-23. Epub 2023 Jul 3. J Virol. 2023. PMID: 37395647 Free PMC article.
-
Pacmanvirus S19, the Second Pacmanvirus Isolated from Sewage Waters in Oran, Algeria.Microbiol Resour Announc. 2021 Oct 21;10(42):e0069321. doi: 10.1128/MRA.00693-21. Epub 2021 Oct 21. Microbiol Resour Announc. 2021. PMID: 34672704 Free PMC article.
-
Asfarviruses and Closely Related Giant Viruses.Viruses. 2023 Apr 20;15(4):1015. doi: 10.3390/v15041015. Viruses. 2023. PMID: 37112995 Free PMC article. Review.
References
-
- Walker P.J., Siddell S.G., Lefkowitz E.J., Mushegian A.R., Dempsey D.M., Dutilh B.E., Harrach B., Harrison R.L., Hendrickson R.C., Junglen S., et al. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019) Arch. Virol. 2019;164:2417–2429. doi: 10.1007/s00705-019-04306-w. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
