

Decreased Ecological Resistance of the Gut Microbiota in Response to Clindamycin Challenge in Mice Colonized with the Fungus Candida albicans
- PMID: 33472981
- PMCID: PMC7845615
- DOI: 10.1128/mSphere.00982-20
Decreased Ecological Resistance of the Gut Microbiota in Response to Clindamycin Challenge in Mice Colonized with the Fungus Candida albicans
Abstract
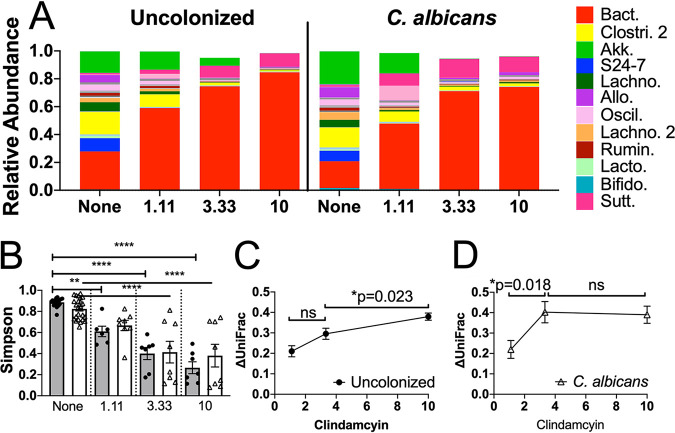
The mammalian gut microbiota is a complex community of microorganisms which typically exhibits remarkable stability. As the gut microbiota has been shown to affect many aspects of host health, the molecular keys to developing and maintaining a "healthy" gut microbiota are highly sought after. Yet, the qualities that define a microbiota as healthy remain elusive. We used the ability to resist change in response to antibiotic disruption, a quality we refer to as ecological resistance, as a metric for the health of the bacterial microbiota. Using a mouse model, we found that colonization with the commensal fungus Candida albicans decreased the ecological resistance of the bacterial microbiota in response to the antibiotic clindamycin such that increased microbiota disruption was observed in C. albicans-colonized mice compared to that in uncolonized mice. C. albicans colonization resulted in decreased alpha diversity and small changes in abundance of bacterial genera prior to clindamycin challenge. Strikingly, co-occurrence network analysis demonstrated that C. albicans colonization resulted in sweeping changes to the co-occurrence network structure, including decreased modularity and centrality and increased density. Thus, C. albicans colonization resulted in changes to the bacterial microbiota community and reduced its ecological resistance.IMPORTANCECandida albicans is the most common fungal member of the human gut microbiota, yet its ability to interact with and affect the bacterial gut microbiota is largely uncharacterized. Previous reports showed limited changes in microbiota composition as defined by bacterial species abundance as a consequence of C. albicans colonization. We also observed only a few bacterial genera that were significantly altered in abundance in C. albicans-colonized mice; however, C. albicans colonization significantly changed the structure of the bacterial microbiota co-occurrence network. Additionally, C. albicans colonization changed the response of the bacterial microbiota ecosystem to a clinically relevant perturbation, challenge with the antibiotic clindamycin.
Keywords: Candida albicans; ecological resistance; microbiota.
Copyright © 2021 Markey et al.
Figures



Similar articles
-
Pre-colonization with the commensal fungus Candida albicans reduces murine susceptibility to Clostridium difficile infection.Gut Microbes. 2018 Nov 2;9(6):497-509. doi: 10.1080/19490976.2018.1465158. Epub 2018 May 30. Gut Microbes. 2018. PMID: 29667487 Free PMC article.
-
Antibiotic-induced gut metabolome and microbiome alterations increase the susceptibility to Candida albicans colonization in the gastrointestinal tract.FEMS Microbiol Ecol. 2020 Jan 1;96(1):fiz187. doi: 10.1093/femsec/fiz187. FEMS Microbiol Ecol. 2020. PMID: 31769789 Free PMC article.
-
Microbiome Resilience despite a Profound Loss of Minority Microbiota following Clindamycin Challenge in Humanized Gnotobiotic Mice.Microbiol Spectr. 2022 Feb 23;10(1):e0196021. doi: 10.1128/spectrum.01960-21. Epub 2022 Jan 12. Microbiol Spectr. 2022. PMID: 35019780 Free PMC article.
-
Candida albicans dwelling in the mammalian gut.Curr Opin Microbiol. 2019 Dec;52:41-46. doi: 10.1016/j.mib.2019.04.007. Epub 2019 May 25. Curr Opin Microbiol. 2019. PMID: 31132744 Review.
-
The gut, the bad and the harmless: Candida albicans as a commensal and opportunistic pathogen in the intestine.Curr Opin Microbiol. 2020 Aug;56:7-15. doi: 10.1016/j.mib.2020.05.006. Epub 2020 Jun 27. Curr Opin Microbiol. 2020. PMID: 32604030 Free PMC article. Review.
Cited by
-
Leaky gut biomarkers in casein- and gluten-rich diet fed rat model of autism.Transl Neurosci. 2021 Dec 31;12(1):601-610. doi: 10.1515/tnsci-2020-0207. eCollection 2021 Jan 1. Transl Neurosci. 2021. PMID: 35070443 Free PMC article.
-
The interplay between gut bacteria and the yeast Candida albicans.Gut Microbes. 2021 Jan-Dec;13(1):1979877. doi: 10.1080/19490976.2021.1979877. Gut Microbes. 2021. PMID: 34586038 Free PMC article.
References
-
- Hippe B, Remely M, Bartosiewicz N, Riedel M, Nichterl C, Schatz L, Pummer S, Haslberger A. 2014. Abundance and diversity of GI microbiota rather than IgG4 levels correlate with abdominal inconvenience and gut permeability in consumers claiming food intolerances. Endocr Metab Immune Disord Drug Targets 14:67–75. doi:10.2174/1871530314666140207103335. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
