

Heparanome-Mediated Rescue of Oligodendrocyte Progenitor Quiescence following Inflammatory Demyelination
- PMID: 33472827
- PMCID: PMC8018763
- DOI: 10.1523/JNEUROSCI.0580-20.2021
Heparanome-Mediated Rescue of Oligodendrocyte Progenitor Quiescence following Inflammatory Demyelination
Abstract
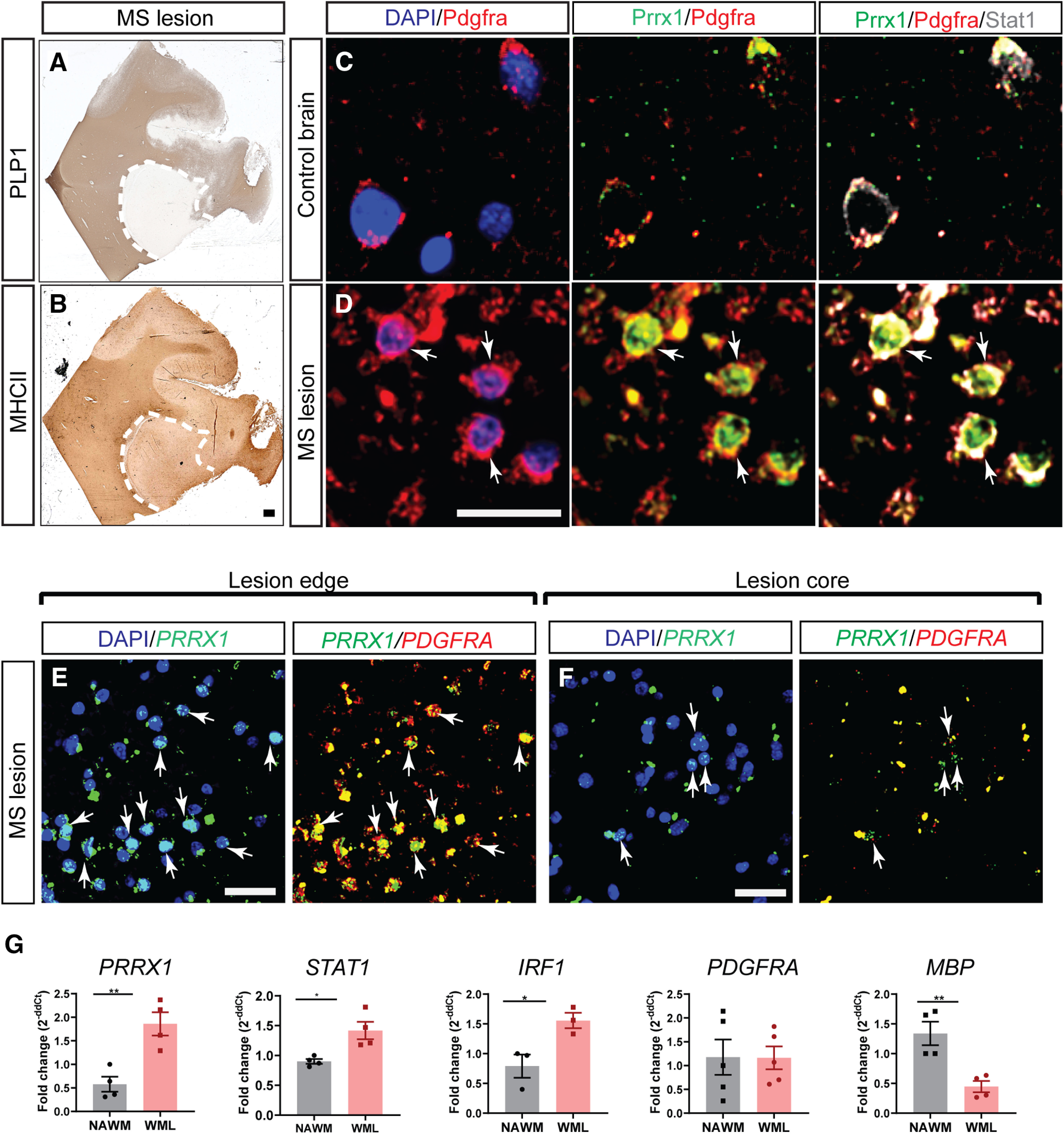
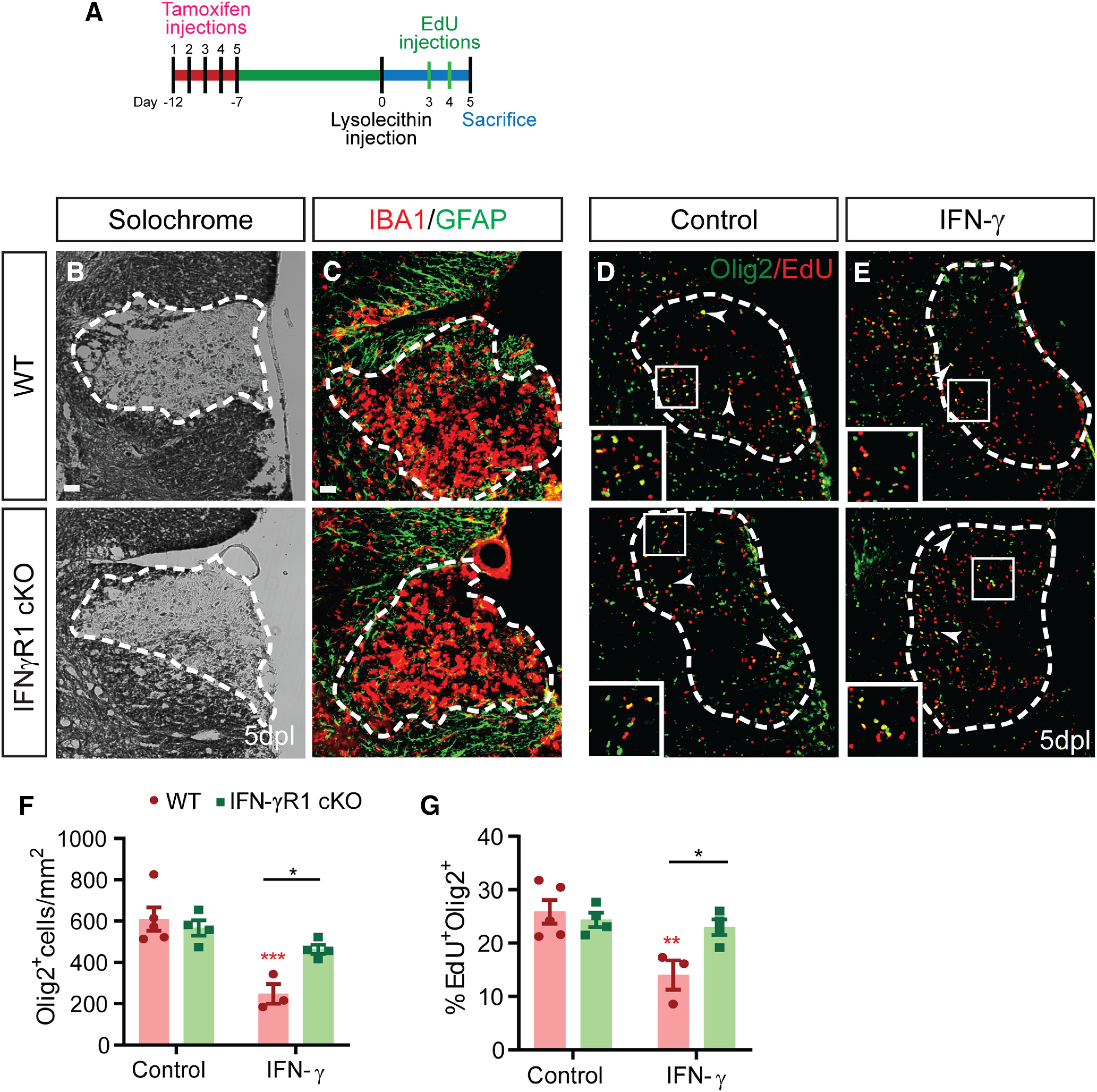
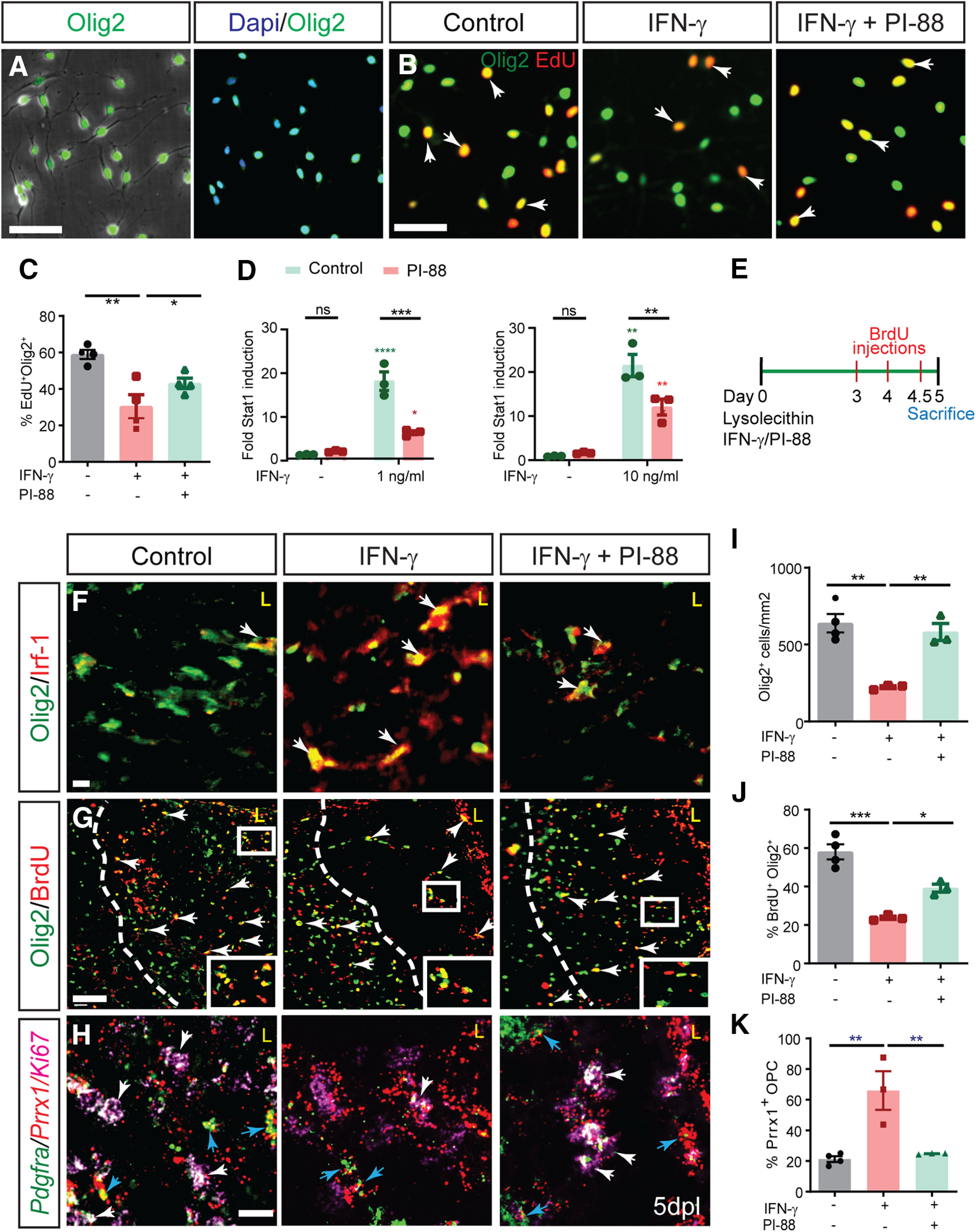
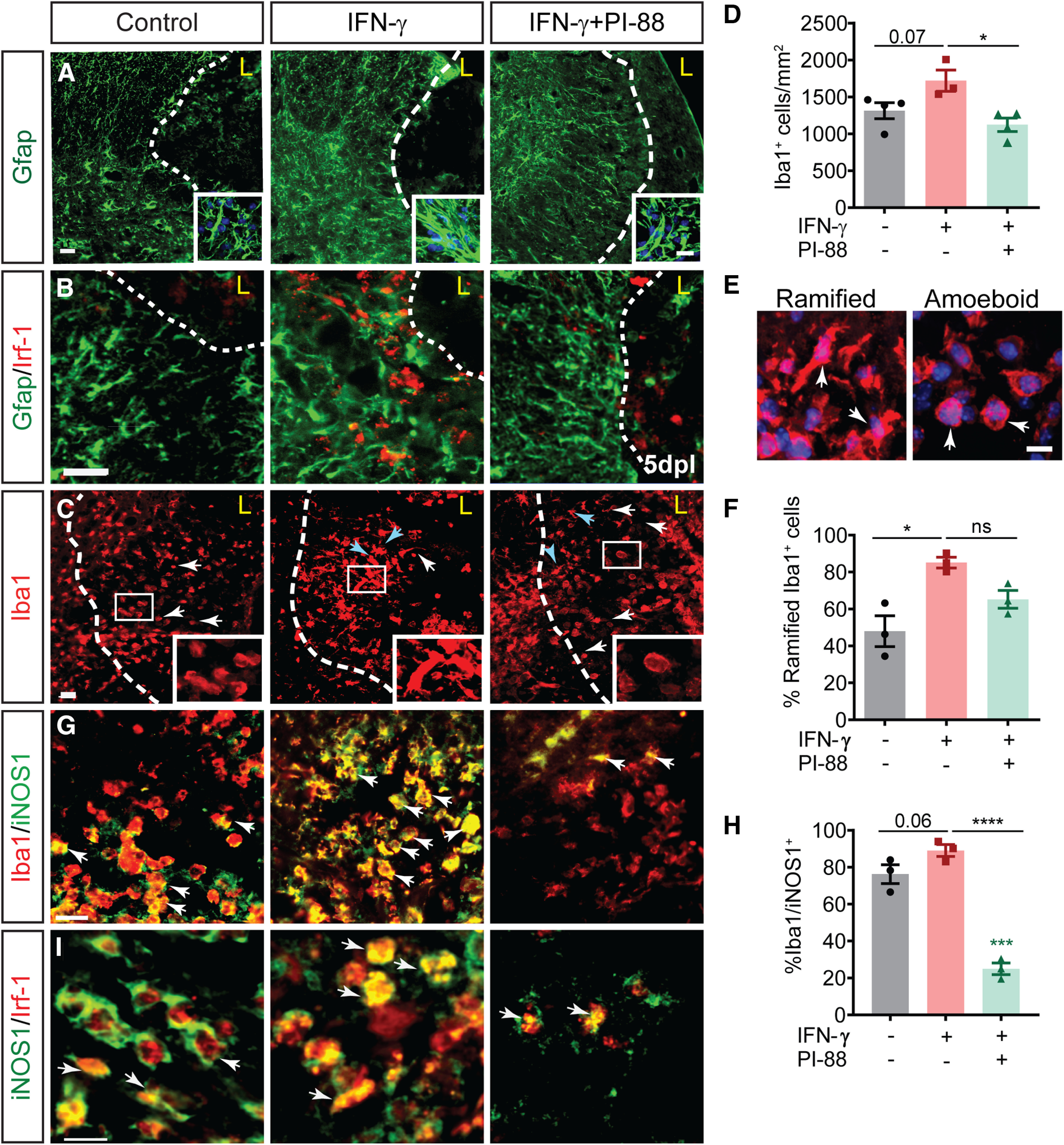
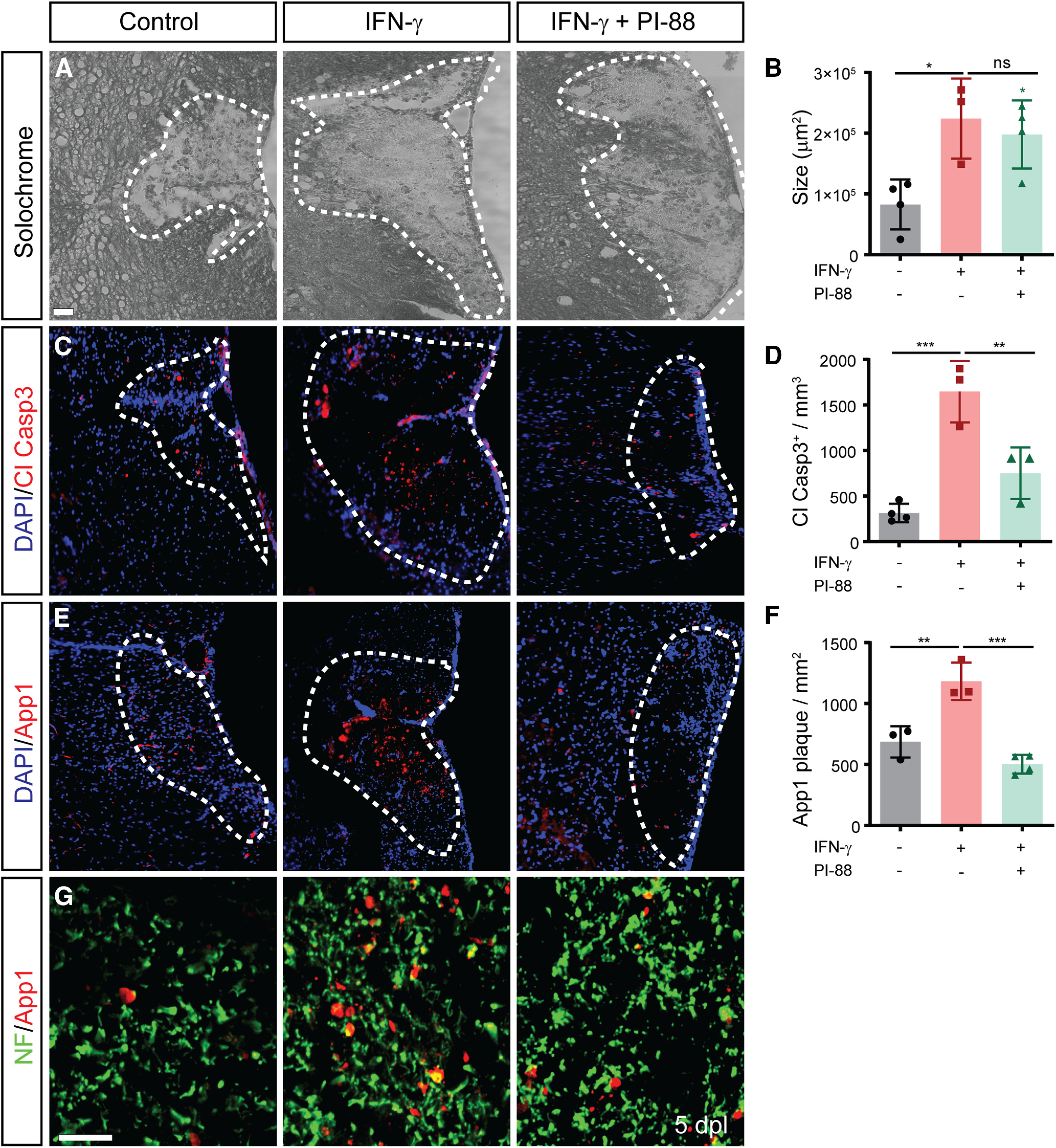
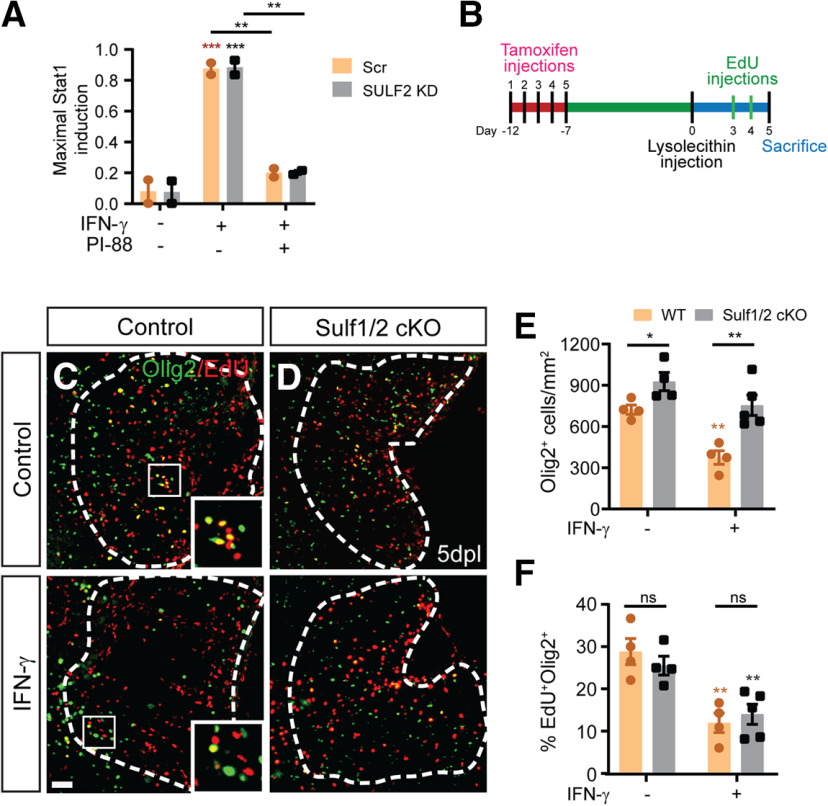
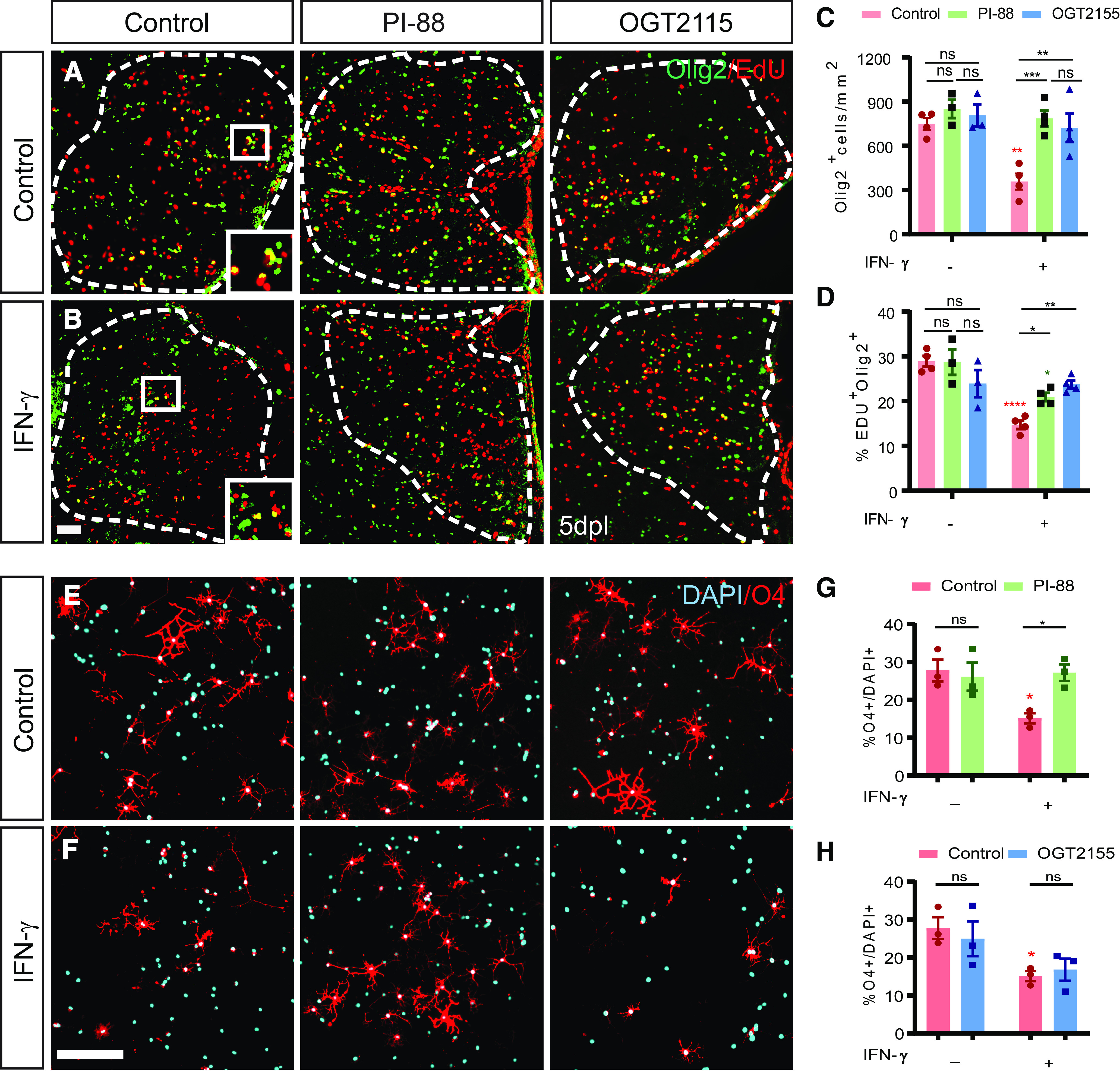
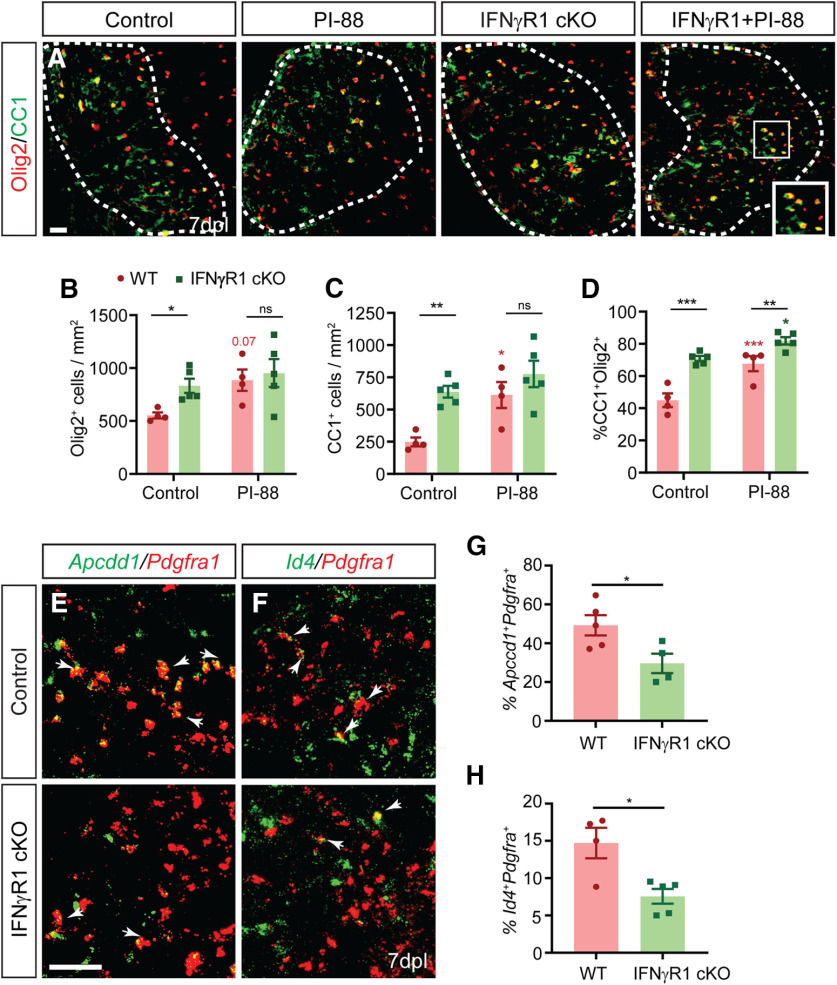
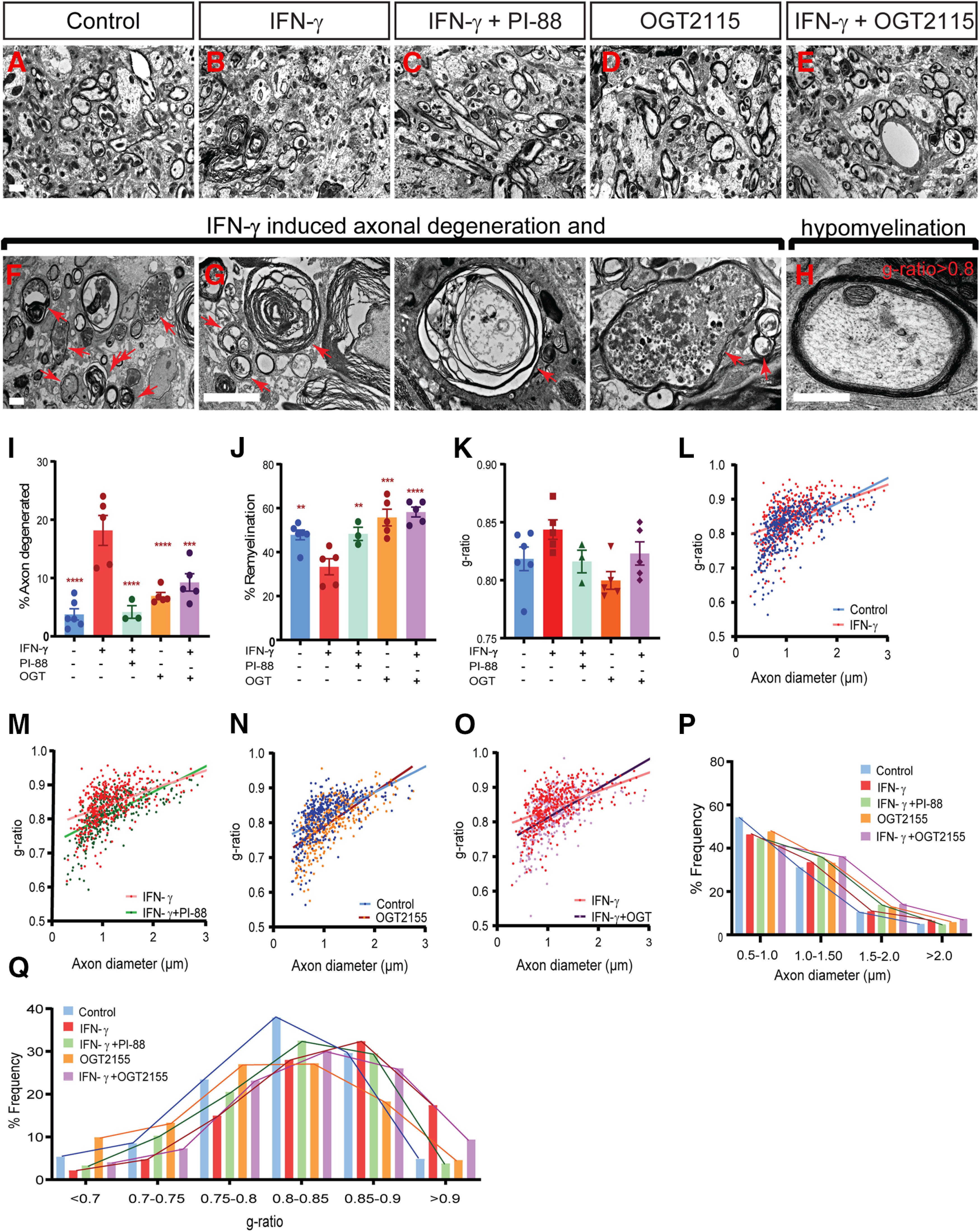
The proinflammatory cytokine IFN-γ, which is chronically elevated in multiple sclerosis, induces pathologic quiescence in human oligodendrocyte progenitor cells (OPCs) via upregulation of the transcription factor PRRX1. In this study using animals of both sexes, we investigated the role of heparan sulfate proteoglycans in the modulation of IFN-γ signaling following demyelination. We found that IFN-γ profoundly impaired OPC proliferation and recruitment following adult spinal cord demyelination. IFN-γ-induced quiescence was mediated by direct signaling in OPCs as conditional genetic ablation of IFNγR1 (Ifngr1) in adult NG2+ OPCs completely abrogated these inhibitory effects. Intriguingly, OPC-specific IFN-γ signaling contributed to failed oligodendrocyte differentiation, which was associated with hyperactive Wnt/Bmp target gene expression in OPCs. We found that PI-88, a heparan sulfate mimetic, directly antagonized IFN-γ to rescue human OPC proliferation and differentiation in vitro and blocked the IFN-γ-mediated inhibitory effects on OPC recruitment in vivo Importantly, heparanase modulation by PI-88 or OGT2155 in demyelinated lesions rescued IFN-γ-mediated axonal damage and demyelination. In addition to OPC-specific effects, IFN-γ-augmented lesions were characterized by increased size, reactive astrogliosis, and proinflammatory microglial/macrophage activation along with exacerbated axonal injury and cell death. Heparanase inhibitor treatment rescued many of the negative IFN-γ-induced sequelae suggesting a profound modulation of the lesion environment. Together, these results suggest that the modulation of the heparanome represents a rational approach to mitigate the negative effects of proinflammatory signaling and rescuing pathologic quiescence in the inflamed and demyelinated human brain.SIGNIFICANCE STATEMENT The failure of remyelination in multiple sclerosis contributes to neurologic dysfunction and neurodegeneration. The activation and proliferation of oligodendrocyte progenitor cells (OPCs) is a necessary step in the recruitment phase of remyelination. Here, we show that the proinflammatory cytokine interferon-γ directly acts on OPCs to induce pathologic quiescence and thereby limit recruitment following demyelination. Heparan sulfate is a highly structured sulfated carbohydrate polymer that is present on the cell surface and regulates several aspects of the signaling microenvironment. We find that pathologic interferon-γ can be blocked by modulation of the heparanome following demyelination using either a heparan mimetic or by treatment with heparanase inhibitor. These studies establish the potential for modulation of heparanome as a regenerative approach in demyelinating disease.
Keywords: demyelination; human; interferon; oligodendrocyte progenitor; quiescence; remyelination.
Copyright © 2021 the authors.
Figures
Similar articles
-
Overcoming the inhibitory microenvironment surrounding oligodendrocyte progenitor cells following experimental demyelination.Nat Commun. 2021 Mar 26;12(1):1923. doi: 10.1038/s41467-021-22263-4. Nat Commun. 2021. PMID: 33772011 Free PMC article.
-
Chronic demyelination of rabbit lesions is attributable to failed oligodendrocyte progenitor cell repopulation.Glia. 2023 Apr;71(4):1018-1035. doi: 10.1002/glia.24324. Epub 2022 Dec 20. Glia. 2023. PMID: 36537341 Free PMC article.
-
Sox2 Sustains Recruitment of Oligodendrocyte Progenitor Cells following CNS Demyelination and Primes Them for Differentiation during Remyelination.J Neurosci. 2015 Aug 19;35(33):11482-99. doi: 10.1523/JNEUROSCI.3655-14.2015. J Neurosci. 2015. PMID: 26290228 Free PMC article.
-
Oligodendrocyte progenitor cell recruitment and remyelination in multiple sclerosis: the more, the merrier?Brain. 2022 Dec 19;145(12):4178-4192. doi: 10.1093/brain/awac307. Brain. 2022. PMID: 36093726 Review.
-
Engineering biomaterial microenvironments to promote myelination in the central nervous system.Brain Res Bull. 2019 Oct;152:159-174. doi: 10.1016/j.brainresbull.2019.07.013. Epub 2019 Jul 12. Brain Res Bull. 2019. PMID: 31306690 Review.
Cited by
-
Quantitative proteomics and multi-omics analysis identifies potential biomarkers and the underlying pathological molecular networks in Chinese patients with multiple sclerosis.BMC Neurol. 2024 Oct 31;24(1):423. doi: 10.1186/s12883-024-03926-3. BMC Neurol. 2024. PMID: 39478468 Free PMC article.
-
Ruxolitinib attenuates secondary injury after traumatic spinal cord injury.Neural Regen Res. 2022 Sep;17(9):2029-2035. doi: 10.4103/1673-5374.335165. Neural Regen Res. 2022. PMID: 35142693 Free PMC article.
-
The interplay of inflammation and remyelination: rethinking MS treatment with a focus on oligodendrocyte progenitor cells.Mol Neurodegener. 2024 Jul 12;19(1):53. doi: 10.1186/s13024-024-00742-8. Mol Neurodegener. 2024. PMID: 38997755 Free PMC article. Review.
-
Unlocking the Potential: immune functions of oligodendrocyte precursor cells.Front Immunol. 2024 Jul 9;15:1425706. doi: 10.3389/fimmu.2024.1425706. eCollection 2024. Front Immunol. 2024. PMID: 39044821 Free PMC article. Review.
-
Oligodendrocytes in central nervous system diseases: the effect of cytokine regulation.Neural Regen Res. 2024 Oct 1;19(10):2132-2143. doi: 10.4103/1673-5374.392854. Epub 2024 Jan 8. Neural Regen Res. 2024. PMID: 38488548 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous