

Discovery and optimization of a novel anti-GUCY2c x CD3 bispecific antibody for the treatment of solid tumors
- PMID: 33459147
- PMCID: PMC7833764
- DOI: 10.1080/19420862.2020.1850395
Discovery and optimization of a novel anti-GUCY2c x CD3 bispecific antibody for the treatment of solid tumors
Abstract
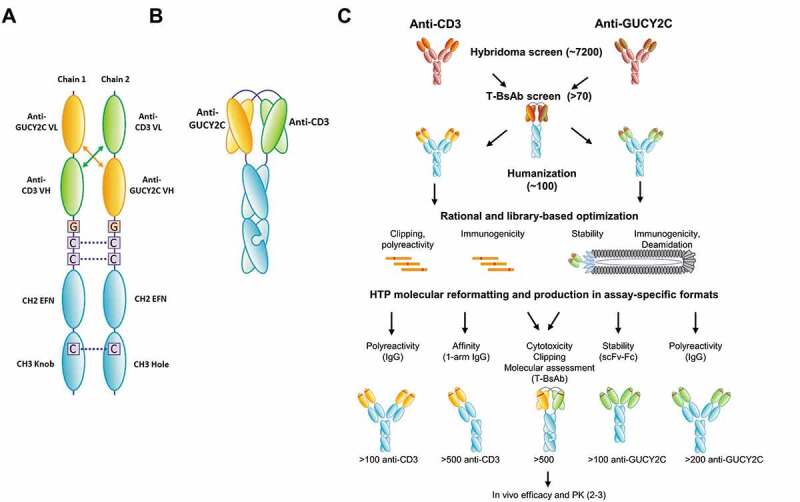
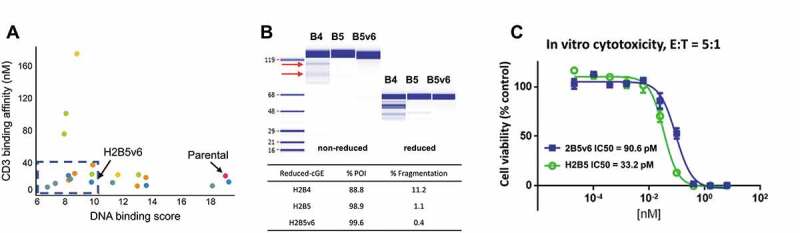
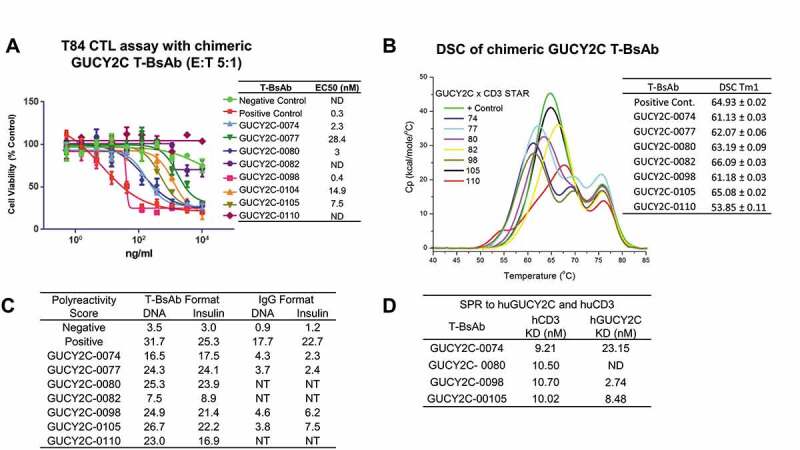
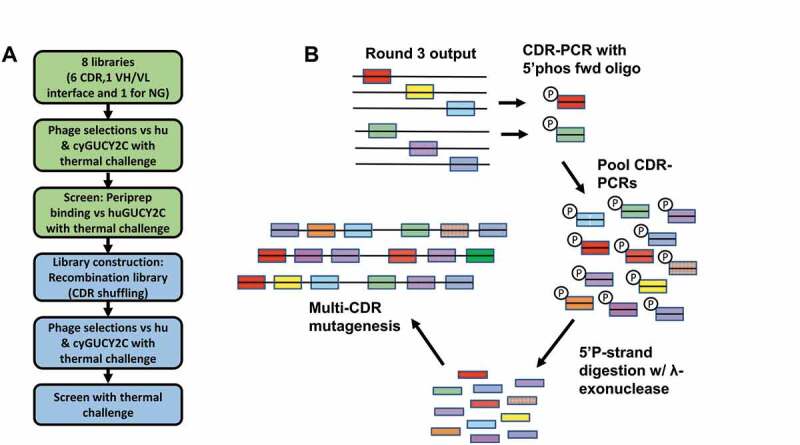
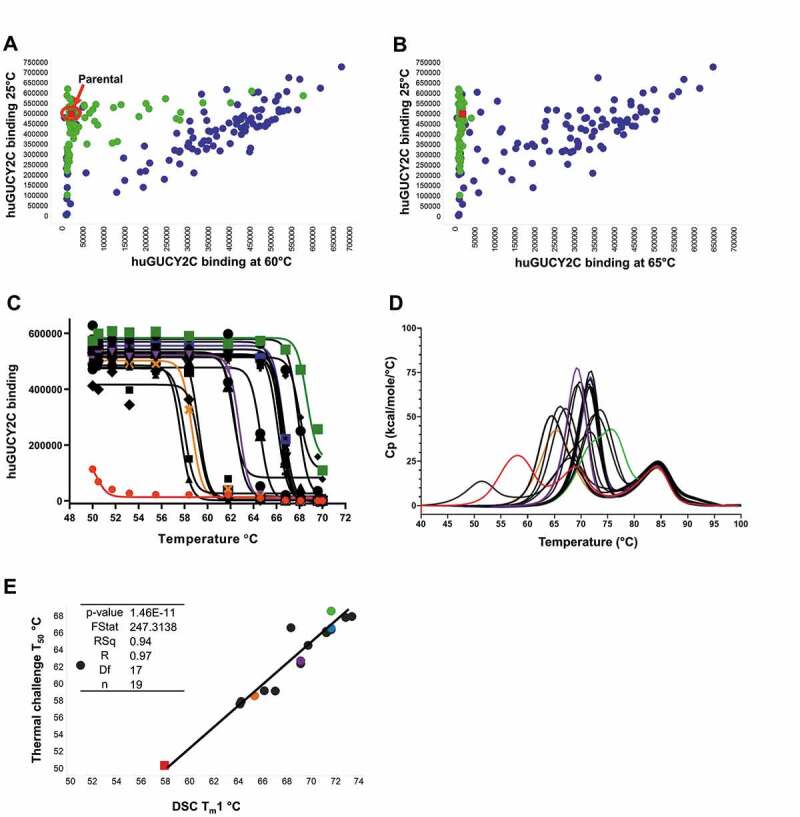
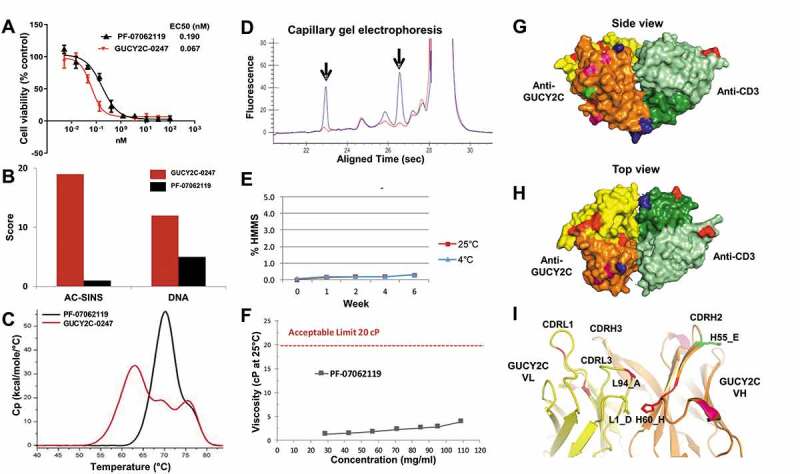
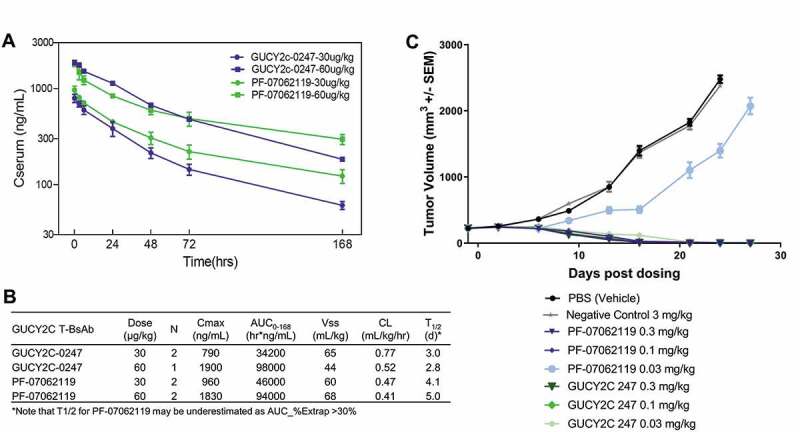
We report here the discovery and optimization of a novel T cell retargeting anti-GUCY2C x anti-CD3ε bispecific antibody for the treatment of solid tumors. Using a combination of hybridoma, phage display and rational design protein engineering, we have developed a fully humanized and manufacturable CD3 bispecific antibody that demonstrates favorable pharmacokinetic properties and potent in vivo efficacy. Anti-GUCY2C and anti-CD3ε antibodies derived from mouse hybridomas were first humanized into well-behaved human variable region frameworks with full retention of binding and T-cell mediated cytotoxic activity. To address potential manufacturability concerns, multiple approaches were taken in parallel to optimize and de-risk the two antibody variable regions. These approaches included structure-guided rational mutagenesis and phage display-based optimization, focusing on improving stability, reducing polyreactivity and self-association potential, removing chemical liabilities and proteolytic cleavage sites, and de-risking immunogenicity. Employing rapid library construction methods as well as automated phage display and high-throughput protein production workflows enabled efficient generation of an optimized bispecific antibody with desirable manufacturability properties, high stability, and low nonspecific binding. Proteolytic cleavage and deamidation in complementarity-determining regions were also successfully addressed. Collectively, these improvements translated to a molecule with potent single-agent in vivo efficacy in a tumor cell line adoptive transfer model and a cynomolgus monkey pharmacokinetic profile (half-life>4.5 days) suitable for clinical development. Clinical evaluation of PF-07062119 is ongoing.
Keywords: GUCY2C; Guanlyate cyclase 2c (GCC); PF-07062119; T cell bispecific; T cell retargeting; T-BsAb; antibody engineering; antibody optimization; developability; high-throughput protein production; immuno-oncology.
Figures
Similar articles
-
A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers.Clin Cancer Res. 2020 May 1;26(9):2188-2202. doi: 10.1158/1078-0432.CCR-19-3275. Epub 2020 Jan 29. Clin Cancer Res. 2020. PMID: 31996389
-
Design, selection and optimization of an anti-TRAIL-R2/anti-CD3 bispecific antibody able to educate T cells to recognize and destroy cancer cells.MAbs. 2018 Oct;10(7):1084-1097. doi: 10.1080/19420862.2018.1494105. Epub 2018 Aug 6. MAbs. 2018. PMID: 29993310 Free PMC article.
-
Single cell-produced and in vitro-assembled anti-FcRH5/CD3 T-cell dependent bispecific antibodies have similar in vitro and in vivo properties.MAbs. 2019 Feb/Mar;11(2):422-433. doi: 10.1080/19420862.2018.1551676. Epub 2018 Dec 17. MAbs. 2019. PMID: 30550367 Free PMC article.
-
[Bispecific antibodies: An old story with a bright future… with CAR-T cells!].Bull Cancer. 2021 Oct;108(10S):S168-S180. doi: 10.1016/j.bulcan.2021.02.016. Bull Cancer. 2021. PMID: 34920800 Review. French.
-
Engineering Immune Cells for in vivo Secretion of Tumor-Specific T Cell-Redirecting Bispecific Antibodies.Front Immunol. 2020 Aug 13;11:1792. doi: 10.3389/fimmu.2020.01792. eCollection 2020. Front Immunol. 2020. PMID: 32903593 Free PMC article. Review.
Cited by
-
Progress on Phage Display Technology: Tailoring Antibodies for Cancer Immunotherapy.Viruses. 2023 Sep 9;15(9):1903. doi: 10.3390/v15091903. Viruses. 2023. PMID: 37766309 Free PMC article. Review.
-
Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics.Antib Ther. 2022 Nov 11;6(1):13-29. doi: 10.1093/abt/tbac029. eCollection 2023 Jan. Antib Ther. 2022. PMID: 36683767 Free PMC article. Review.
-
Molecular insights into recognition of GUCY2C by T-cell engaging bispecific antibody anti-GUCY2CxCD3.Sci Rep. 2023 Aug 17;13(1):13408. doi: 10.1038/s41598-023-40467-0. Sci Rep. 2023. PMID: 37591971 Free PMC article.
-
Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics.Antibodies (Basel). 2021 Mar 30;10(2):13. doi: 10.3390/antib10020013. Antibodies (Basel). 2021. PMID: 33808165 Free PMC article. Review.
-
Design and engineering of bispecific antibodies: insights and practical considerations.Front Bioeng Biotechnol. 2024 Jan 25;12:1352014. doi: 10.3389/fbioe.2024.1352014. eCollection 2024. Front Bioeng Biotechnol. 2024. PMID: 38333084 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources