

Cellular and molecular pathobiology of heart failure with preserved ejection fraction
- PMID: 33432192
- PMCID: PMC8574228
- DOI: 10.1038/s41569-020-00480-6
Cellular and molecular pathobiology of heart failure with preserved ejection fraction
Erratum in
-
Publisher Correction: Cellular and molecular pathobiology of heart failure with preserved ejection fraction.Nat Rev Cardiol. 2021 Oct;18(10):735. doi: 10.1038/s41569-021-00516-5. Nat Rev Cardiol. 2021. PMID: 33479518 No abstract available.
Abstract
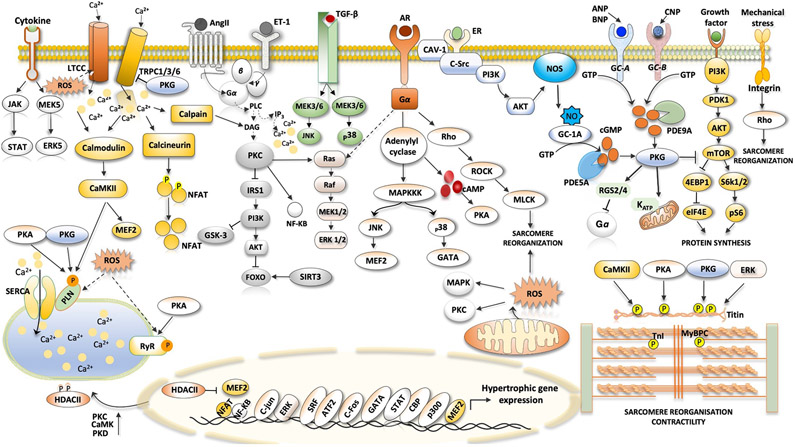
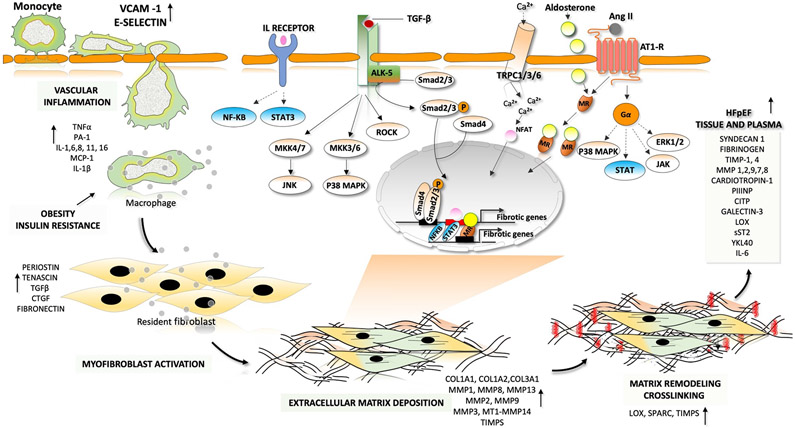
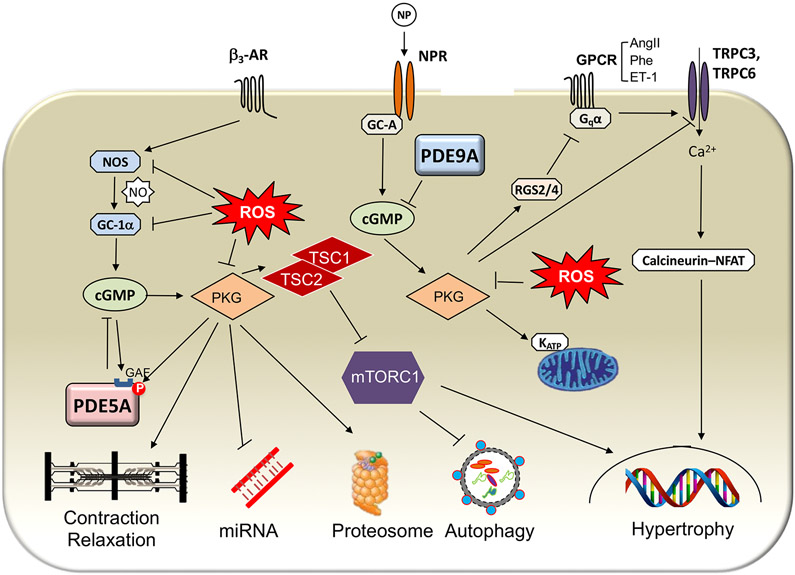
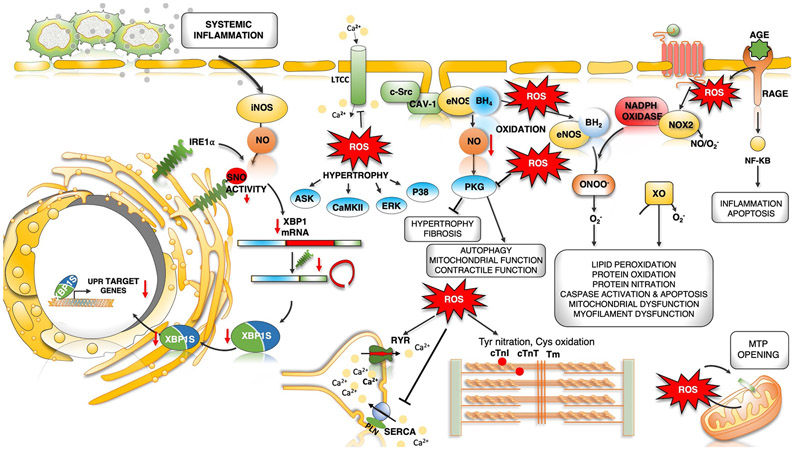
Heart failure with preserved ejection fraction (HFpEF) affects half of all patients with heart failure worldwide, is increasing in prevalence, confers substantial morbidity and mortality, and has very few effective treatments. HFpEF is arguably the greatest unmet medical need in cardiovascular disease. Although HFpEF was initially considered to be a haemodynamic disorder characterized by hypertension, cardiac hypertrophy and diastolic dysfunction, the pandemics of obesity and diabetes mellitus have modified the HFpEF syndrome, which is now recognized to be a multisystem disorder involving the heart, lungs, kidneys, skeletal muscle, adipose tissue, vascular system, and immune and inflammatory signalling. This multiorgan involvement makes HFpEF difficult to model in experimental animals because the condition is not simply cardiac hypertrophy and hypertension with abnormal myocardial relaxation. However, new animal models involving both haemodynamic and metabolic disease, and increasing efforts to examine human pathophysiology, are revealing new signalling pathways and potential therapeutic targets. In this Review, we discuss the cellular and molecular pathobiology of HFpEF, with the major focus being on mechanisms relevant to the heart, because most research has focused on this organ. We also highlight the involvement of other important organ systems, including the lungs, kidneys and skeletal muscle, efforts to characterize patients with the use of systemic biomarkers, and ongoing therapeutic efforts. Our objective is to provide a roadmap of the signalling pathways and mechanisms of HFpEF that are being characterized and which might lead to more patient-specific therapies and improved clinical outcomes.
Figures
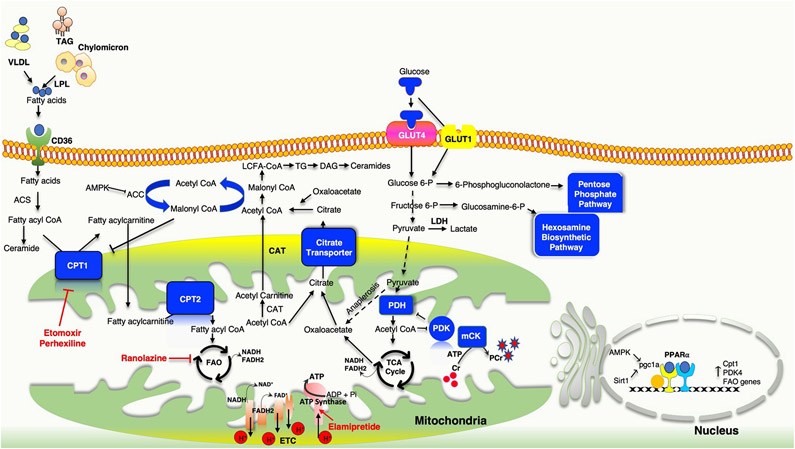
Similar articles
-
Heart failure with preserved ejection fraction.Nat Rev Dis Primers. 2024 Aug 14;10(1):55. doi: 10.1038/s41572-024-00540-y. Nat Rev Dis Primers. 2024. PMID: 39143132 Review.
-
Cardiomyocyte Functional Etiology in Heart Failure With Preserved Ejection Fraction Is Distinctive-A New Preclinical Model.J Am Heart Assoc. 2018 Jun 1;7(11):e007451. doi: 10.1161/JAHA.117.007451. J Am Heart Assoc. 2018. PMID: 29858360 Free PMC article.
-
The aging heart in focus: The advanced understanding of heart failure with preserved ejection fraction.Ageing Res Rev. 2024 Nov;101:102542. doi: 10.1016/j.arr.2024.102542. Epub 2024 Oct 12. Ageing Res Rev. 2024. PMID: 39396676 Review.
-
A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation.J Am Coll Cardiol. 2013 Jul 23;62(4):263-71. doi: 10.1016/j.jacc.2013.02.092. Epub 2013 May 15. J Am Coll Cardiol. 2013. PMID: 23684677 Review.
-
Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap.Circulation. 2016 Jul 5;134(1):73-90. doi: 10.1161/CIRCULATIONAHA.116.021884. Circulation. 2016. PMID: 27358439 Free PMC article. Review.
Cited by
-
Mouse Model of Heart Failure With Preserved Ejection Fraction Driven by Hyperlipidemia and Enhanced Cardiac Low-Density Lipoprotein Receptor Expression.J Am Heart Assoc. 2022 Sep 6;11(17):e027216. doi: 10.1161/JAHA.122.027216. Epub 2022 Sep 3. J Am Heart Assoc. 2022. PMID: 36056728 Free PMC article.
-
Interactions between the gut microbiome, associated metabolites and the manifestation and progression of heart failure with preserved ejection fraction in ZSF1 rats.Cardiovasc Diabetol. 2024 Aug 14;23(1):299. doi: 10.1186/s12933-024-02398-6. Cardiovasc Diabetol. 2024. PMID: 39143579 Free PMC article.
-
The promise of RNA-based therapeutics in revolutionizing heart failure management - a narrative review of current evidence.Ann Med Surg (Lond). 2023 Jul 31;85(9):4442-4453. doi: 10.1097/MS9.0000000000001118. eCollection 2023 Sep. Ann Med Surg (Lond). 2023. PMID: 37663746 Free PMC article. Review.
-
Value of the HFA-PEFF diagnostic algorithms for heart failure with preserved ejection fraction to the inflammatory myopathy population.Arthritis Res Ther. 2023 Aug 4;25(1):141. doi: 10.1186/s13075-023-03131-6. Arthritis Res Ther. 2023. PMID: 37542301 Free PMC article.
-
Catestatin Protects Against Diastolic Dysfunction by Attenuating Mitochondrial Reactive Oxygen Species Generation.J Am Heart Assoc. 2023 May 2;12(9):e029470. doi: 10.1161/JAHA.123.029470. Epub 2023 Apr 29. J Am Heart Assoc. 2023. PMID: 37119063 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
