

Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury
- PMID: 33402181
- PMCID: PMC7787000
- DOI: 10.1186/s12974-020-02041-7
Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury
Abstract
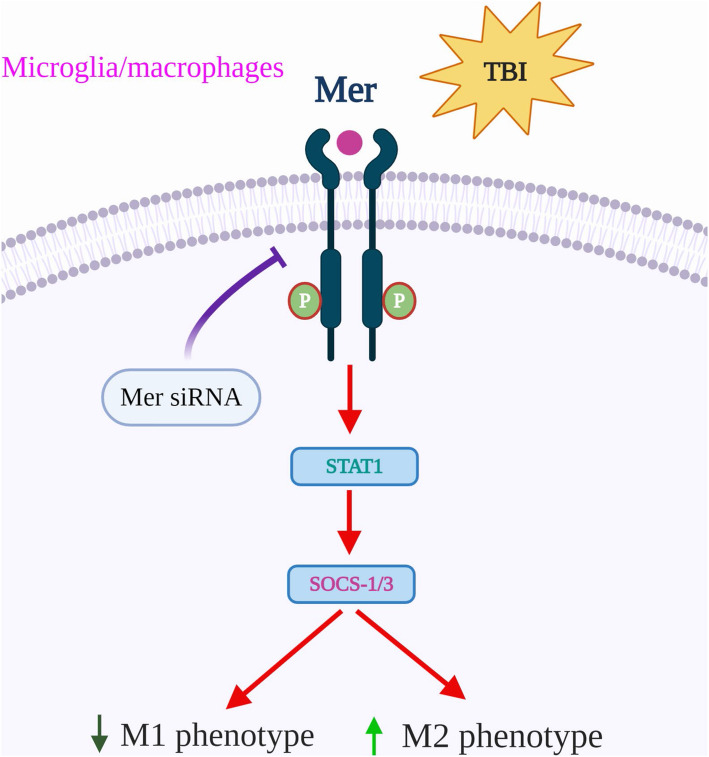
Background: Traumatic brain injury (TBI) is a leading cause of death and disability worldwide. Microglial/macrophage activation and neuroinflammation are key cellular events following TBI, but the regulatory and functional mechanisms are still not well understood. Myeloid-epithelial-reproductive tyrosine kinase (Mer), a member of the Tyro-Axl-Mer (TAM) family of receptor tyrosine kinases, regulates multiple features of microglial/macrophage physiology. However, its function in regulating the innate immune response and microglial/macrophage M1/M2 polarization in TBI has not been addressed. The present study aimed to evaluate the role of Mer in regulating microglial/macrophage M1/M2 polarization and neuroinflammation following TBI.
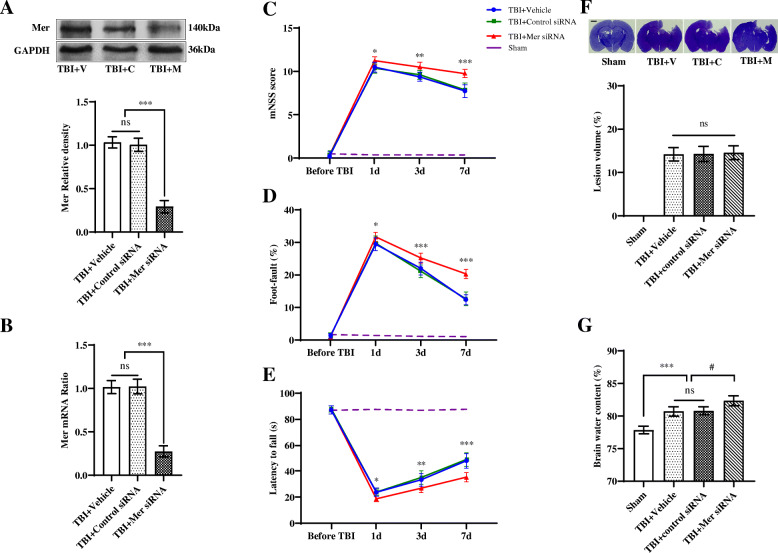
Methods: The controlled cortical impact (CCI) mouse model was employed. Mer siRNA was intracerebroventricularly administered, and recombinant protein S (PS) was intravenously applied for intervention. The neurobehavioral assessments, RT-PCR, Western blot, magnetic-activated cell sorting, immunohistochemistry and confocal microscopy analysis, Nissl and Fluoro-Jade B staining, brain water content measurement, and contusion volume assessment were performed.
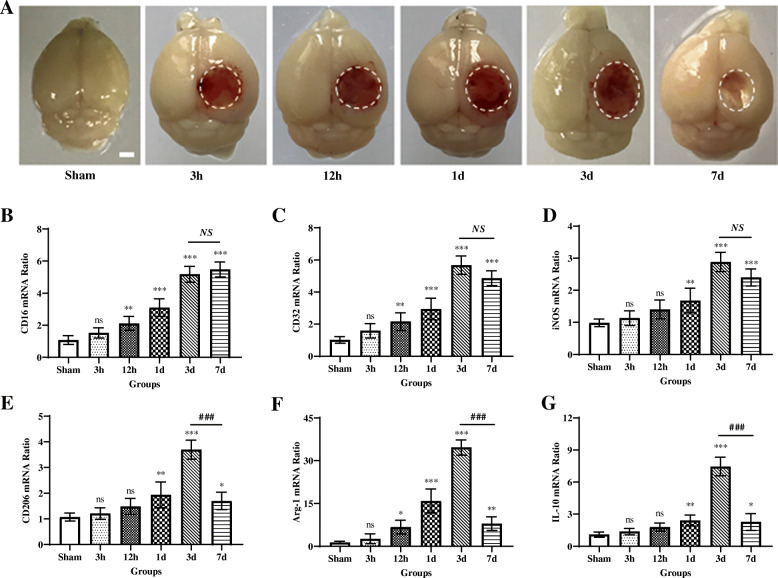
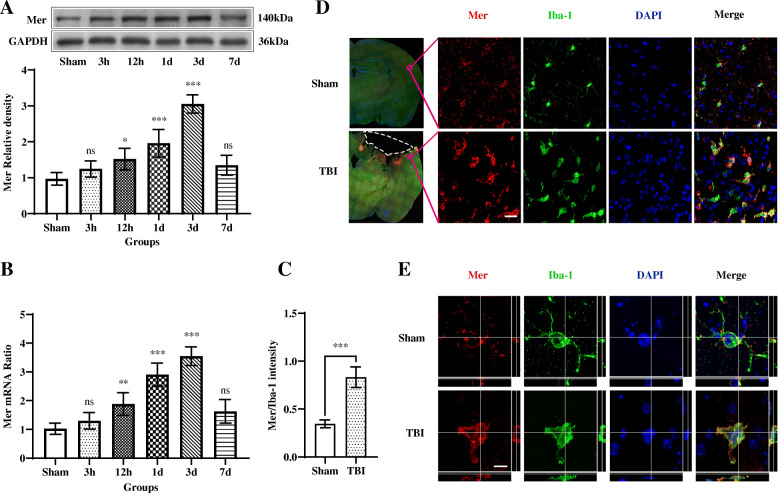
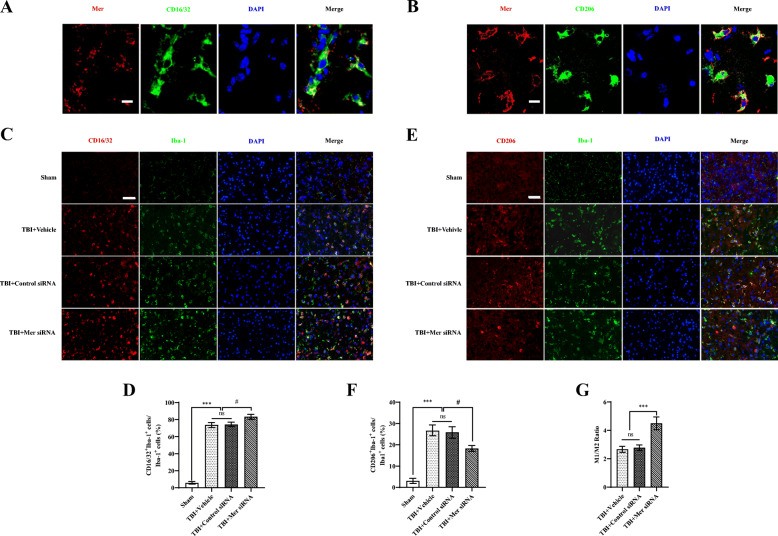
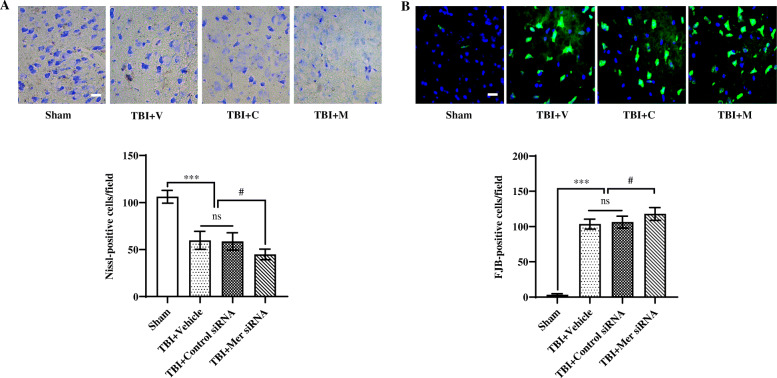
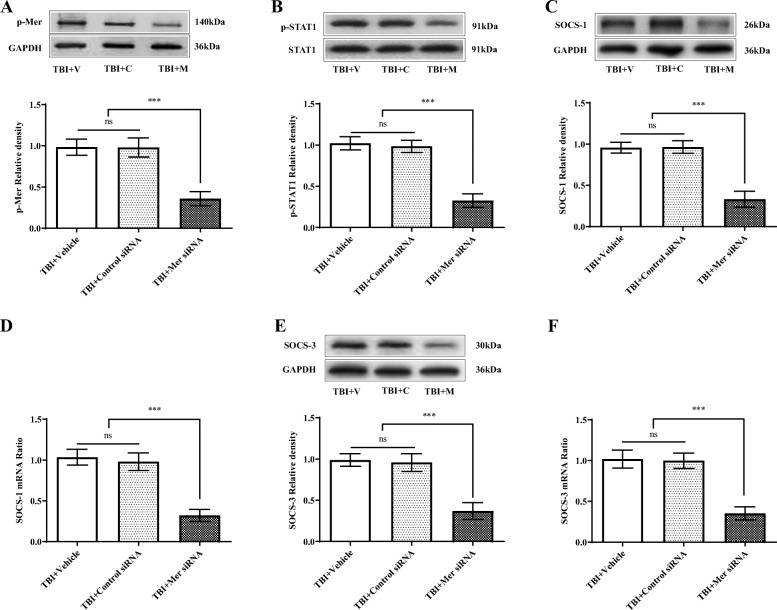
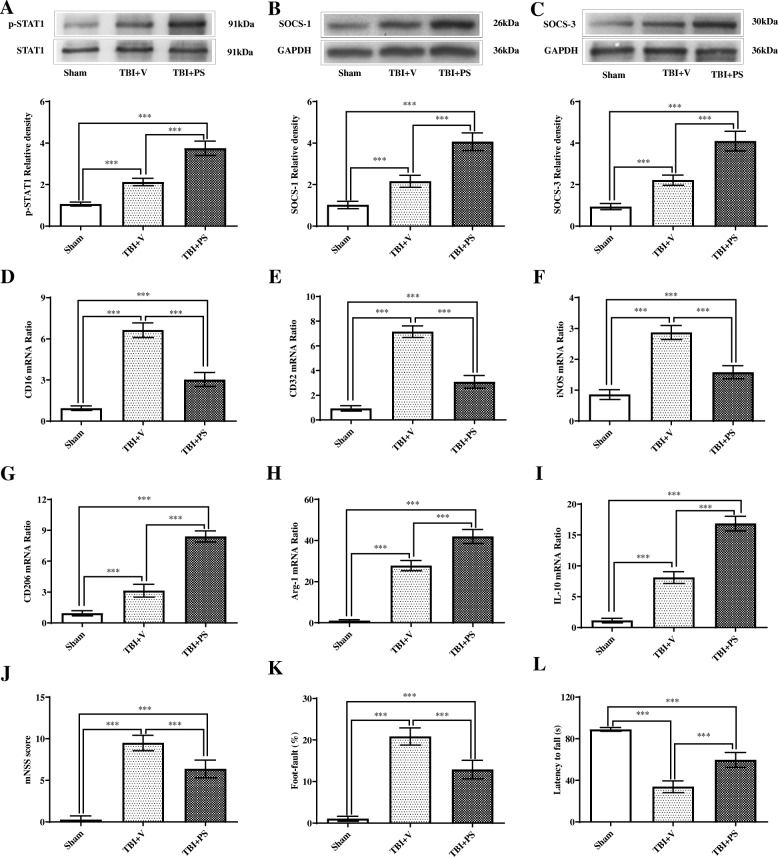
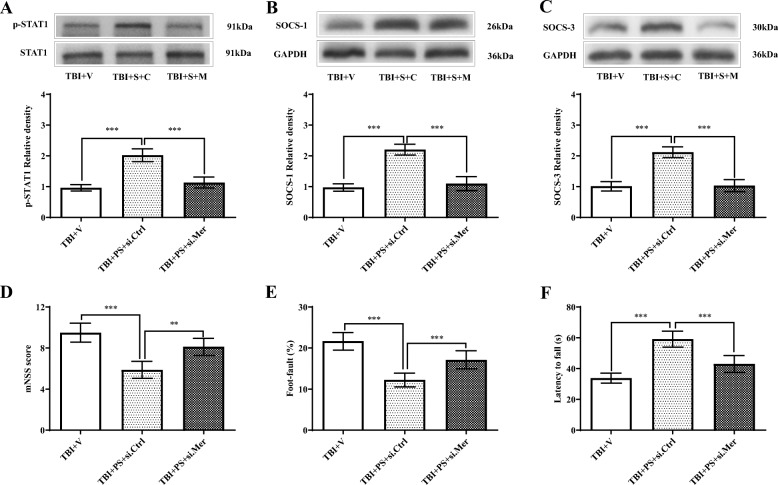
Results: Mer is upregulated and regulates microglial/macrophage M1/M2 polarization and neuroinflammation in the acute stage of TBI. Mechanistically, Mer activates the signal transducer and activator of transcription 1 (STAT1)/suppressor of cytokine signaling 1/3 (SOCS1/3) pathway. Inhibition of Mer markedly decreases microglial/macrophage M2-like polarization while increases M1-like polarization, which exacerbates the secondary brain damage and sensorimotor deficits after TBI. Recombinant PS exerts beneficial effects in TBI mice through Mer activation.
Conclusions: Mer is an important regulator of microglial/macrophage M1/M2 polarization and neuroinflammation, and may be considered as a potential target for therapeutic intervention in TBI.
Keywords: M1/M2 polarization; Mer; Microglia/macrophage; Neuroinflammation; TBI.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury.Brain Behav Immun. 2016 Nov;58:291-309. doi: 10.1016/j.bbi.2016.07.158. Epub 2016 Jul 28. Brain Behav Immun. 2016. PMID: 27477920 Free PMC article.
-
Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury.J Neuroinflammation. 2018 Apr 20;15(1):116. doi: 10.1186/s12974-018-1151-3. J Neuroinflammation. 2018. PMID: 29678169 Free PMC article.
-
Mer activation ameliorates nerve injury-induced neuropathic pain by regulating microglial polarization and neuroinflammation via SOCS3 in male rats.Naunyn Schmiedebergs Arch Pharmacol. 2024 Sep;397(9):7037-7050. doi: 10.1007/s00210-024-03070-2. Epub 2024 Apr 19. Naunyn Schmiedebergs Arch Pharmacol. 2024. PMID: 38639897
-
The Polarization States of Microglia in TBI: A New Paradigm for Pharmacological Intervention.Neural Plast. 2017;2017:5405104. doi: 10.1155/2017/5405104. Epub 2017 Feb 1. Neural Plast. 2017. PMID: 28255460 Free PMC article. Review.
-
Microglia in the TBI brain: The good, the bad, and the dysregulated.Exp Neurol. 2016 Jan;275 Pt 3(0 3):316-327. doi: 10.1016/j.expneurol.2015.08.018. Epub 2015 Sep 3. Exp Neurol. 2016. PMID: 26342753 Free PMC article. Review.
Cited by
-
Cognitive Deficits Found in a Pro-inflammatory State are Independent of ERK1/2 Signaling in the Murine Brain Hippocampus Treated with Shiga Toxin 2 from Enterohemorrhagic Escherichia coli.Cell Mol Neurobiol. 2023 Jul;43(5):2203-2217. doi: 10.1007/s10571-022-01298-1. Epub 2022 Oct 13. Cell Mol Neurobiol. 2023. PMID: 36227397
-
Suppressor of cytokine signaling 3-derived peptide as a therapeutic for inflammatory and oxidative stress-induced damage to the retina.Mol Vis. 2023 Dec 20;29:338-356. eCollection 2023. Mol Vis. 2023. PMID: 38264613 Free PMC article.
-
Calming the Nerves via the Immune Instructive Physiochemical Properties of Self-Assembling Peptide Hydrogels.Adv Sci (Weinh). 2024 Feb;11(5):e2303707. doi: 10.1002/advs.202303707. Epub 2023 Nov 29. Adv Sci (Weinh). 2024. PMID: 38030559 Free PMC article. Review.
-
Gut Microbiota Dysbiosis after Traumatic Brain Injury Contributes to Persistent Microglial Activation Associated with Upregulated Lyz2 and Shifted Tryptophan Metabolic Phenotype.Nutrients. 2022 Aug 24;14(17):3467. doi: 10.3390/nu14173467. Nutrients. 2022. PMID: 36079724 Free PMC article.
-
BAP1 Loss Promotes Suppressive Tumor Immune Microenvironment via Upregulation of PROS1 in Class 2 Uveal Melanomas.Cancers (Basel). 2022 Jul 28;14(15):3678. doi: 10.3390/cancers14153678. Cancers (Basel). 2022. PMID: 35954340 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
