

Protein Tyrosine Phosphatase SHP2 Suppresses Host Innate Immunity against Influenza A Virus by Regulating EGFR-Mediated Signaling
- PMID: 33361428
- PMCID: PMC8094946
- DOI: 10.1128/JVI.02001-20
Protein Tyrosine Phosphatase SHP2 Suppresses Host Innate Immunity against Influenza A Virus by Regulating EGFR-Mediated Signaling
Abstract
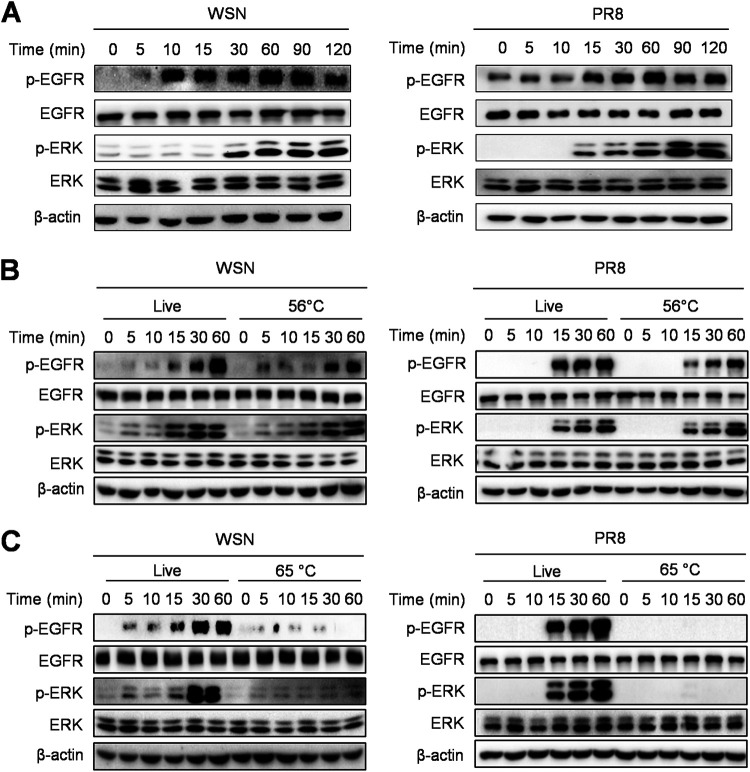
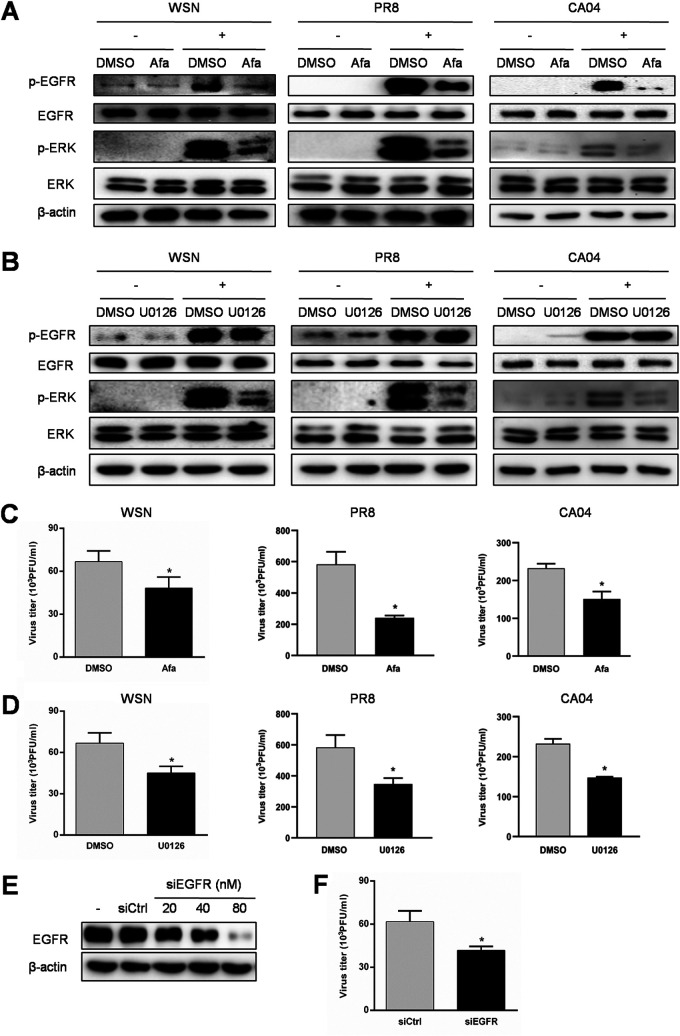
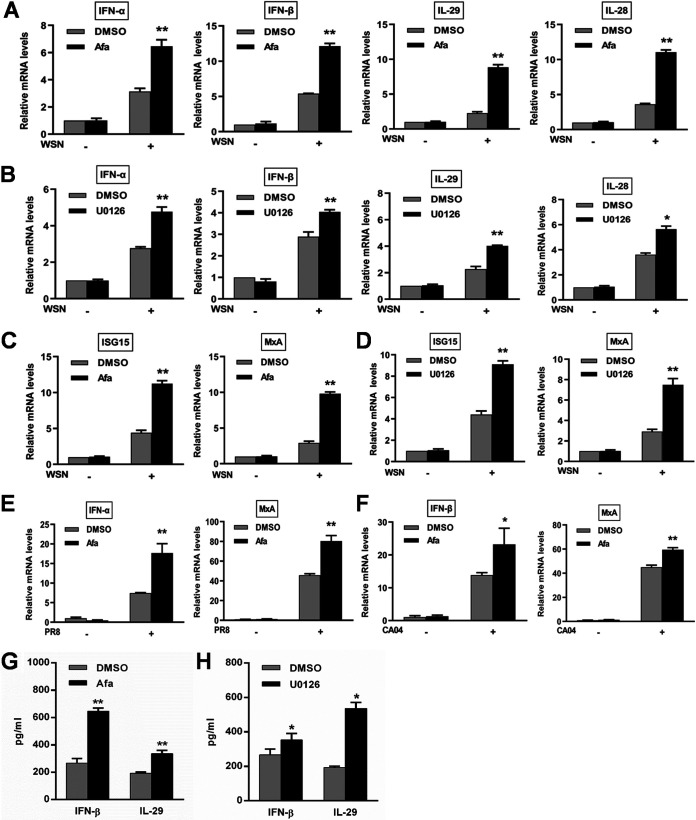
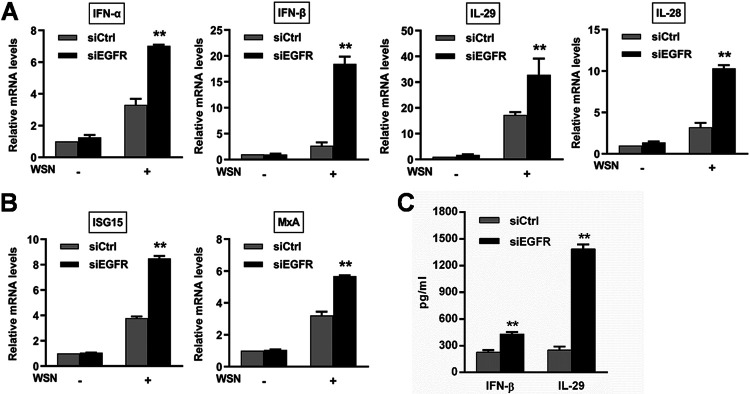
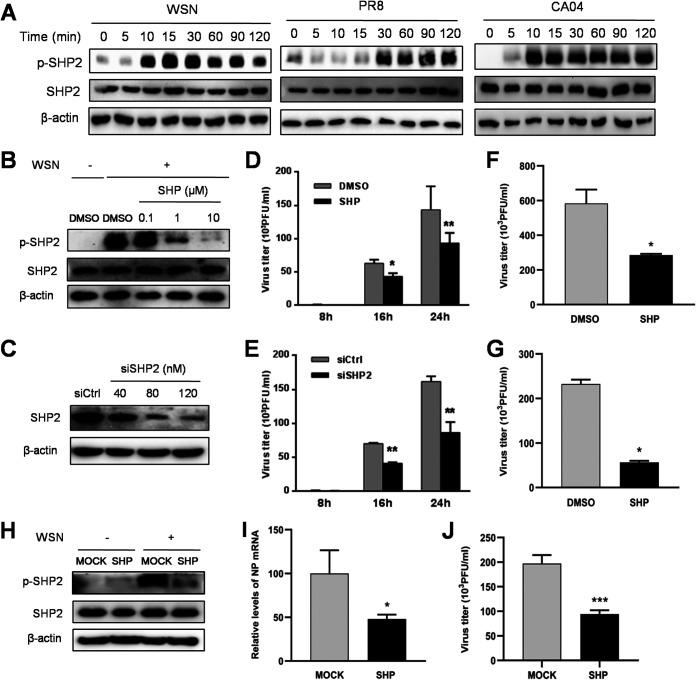
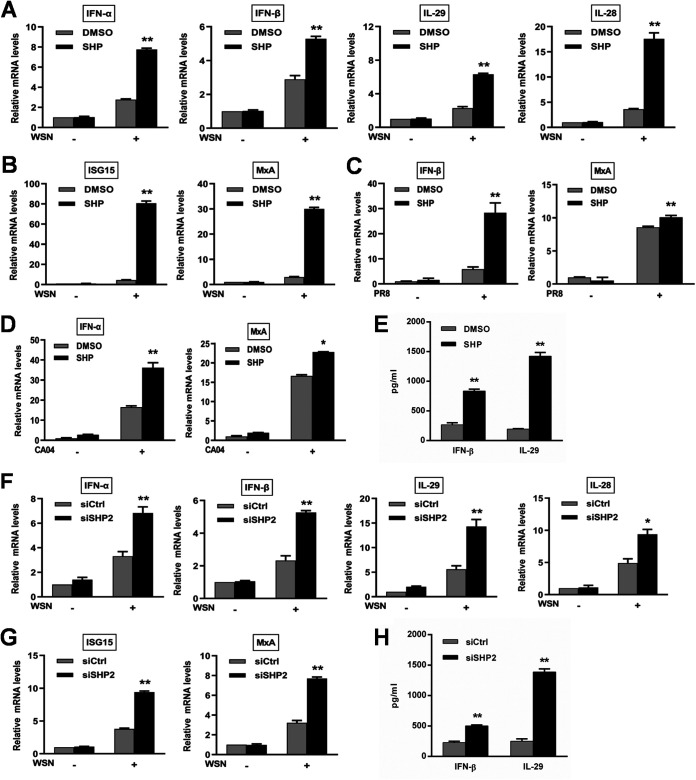
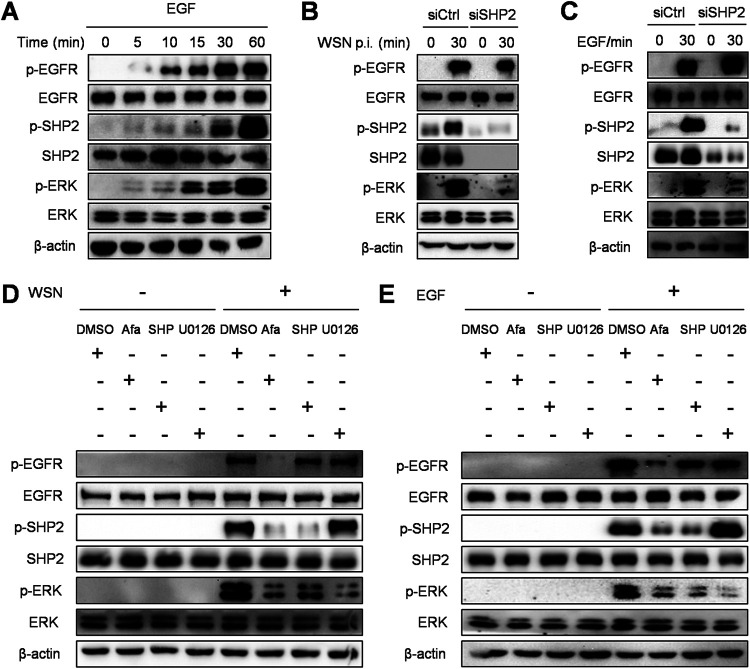
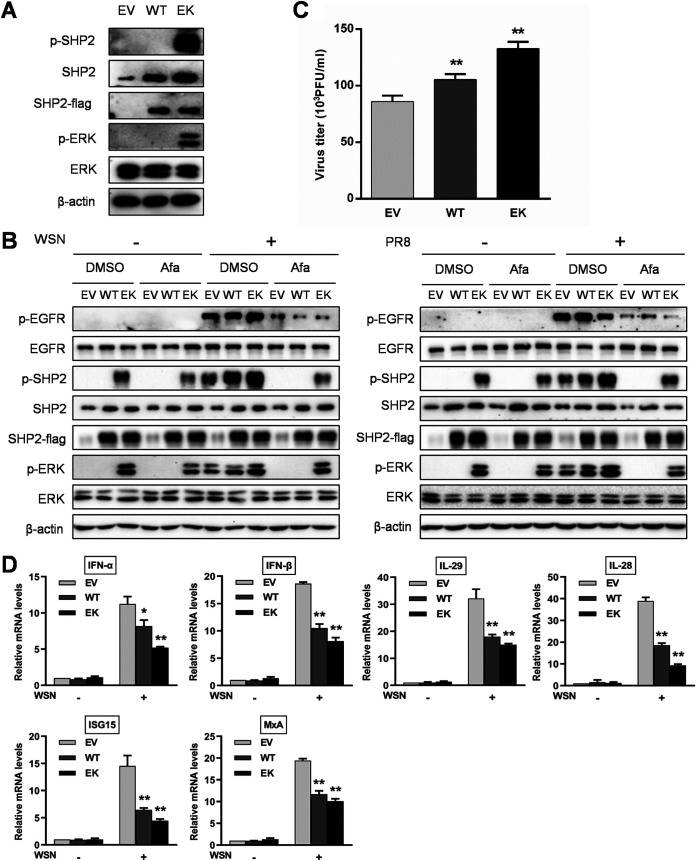
Influenza A virus (IAV) is a highly contagious pathogen, causing acute respiratory illnesses in human beings and animals and frequently giving rise to epidemic outbreaks. Evasion by IAV of host immunity facilitates viral replication and spread, which can be initiated through various mechanisms, including epidermal growth factor receptor (EGFR) activation. However, how EGFR mediates the suppression of antiviral systems remains unclear. Here, we examined host innate immune responses and their relevant signaling to EGFR upon IAV infection. IAV was found to induce the phosphorylation of EGFR and extracellular signal-regulated kinase (ERK) at an early stage of infection. Inhibition of EGFR or ERK suppressed the viral replication but increased the expression of type I and type III interferons (IFNs) and interferon-stimulated genes (ISGs), supporting the idea that IAV escapes from antiviral innate immunity by activating EGFR/ERK signaling. Meanwhile, IAV infection also induced the activation of Src homology region 2-containing protein tyrosine phosphatase 2 (SHP2). Pharmacological inhibition or small interfering RNA (siRNA)-based silencing of SHP2 enhanced the IFN-dependent antiviral activity and reduced virion production. Furthermore, knockdown of SHP2 attenuated the EGFR-mediated ERK phosphorylation triggered by viral infection or EGF stimulation. Conversely, ectopic expression of constitutively active SHP2 noticeably promoted ERK activation and viral replication, concomitant with diminished immune function. Altogether, the results indicate that SHP2 is crucial for IAV-induced activation of the EGFR/ERK pathway to suppress host antiviral responses.IMPORTANCE Viral immune evasion is the most important strategy whereby viruses evolve for their survival. This work shows that influenza A virus (IAV) suppressed the antiviral innate immunity through downregulation of IFNs and ISGs by activating EGFR/ERK signaling. Meanwhile, IAV also induced the activation of protein tyrosine phosphatase SHP2, which was found to be responsible for modulating the EGFR-mediated ERK activity and subsequent antiviral effectiveness both in vitro and in vivo The results suggest that SHP2 is a key signal transducer between EGFR and ERK and plays a crucial role in suppressing host innate immunity during IAV infection. The finding enhances our understanding of influenza immune evasion and provides a new therapeutic approach to viral infection.
Keywords: EGFR; SHP2; influenza A virus; innate immunity; interferon.
Copyright © 2021 American Society for Microbiology.
Figures
Similar articles
-
Inducible Guanylate-Binding Protein 7 Facilitates Influenza A Virus Replication by Suppressing Innate Immunity via NF-κB and JAK-STAT Signaling Pathways.J Virol. 2021 Feb 24;95(6):e02038-20. doi: 10.1128/JVI.02038-20. Print 2021 Feb 24. J Virol. 2021. PMID: 33408175 Free PMC article.
-
Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression.J Virol. 2014 Aug;88(15):8375-85. doi: 10.1128/JVI.00126-14. Epub 2014 May 14. J Virol. 2014. PMID: 24829357 Free PMC article.
-
Identification of lncRNA-155 encoded by MIR155HG as a novel regulator of innate immunity against influenza A virus infection.Cell Microbiol. 2019 Aug;21(8):e13036. doi: 10.1111/cmi.13036. Epub 2019 May 29. Cell Microbiol. 2019. PMID: 31045320
-
Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins.Viruses. 2018 Dec 12;10(12):708. doi: 10.3390/v10120708. Viruses. 2018. PMID: 30545063 Free PMC article. Review.
-
The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains?J Innate Immun. 2020;12(6):437-447. doi: 10.1159/000508379. Epub 2020 Jun 19. J Innate Immun. 2020. PMID: 32564033 Free PMC article. Review.
Cited by
-
Metabolomic profiling of Marek's disease virus infection in host cell based on untargeted LC-MS.Front Microbiol. 2023 Nov 9;14:1270762. doi: 10.3389/fmicb.2023.1270762. eCollection 2023. Front Microbiol. 2023. PMID: 38029131 Free PMC article.
-
Virus versus host: influenza A virus circumvents the immune responses.Front Microbiol. 2024 May 16;15:1394510. doi: 10.3389/fmicb.2024.1394510. eCollection 2024. Front Microbiol. 2024. PMID: 38817972 Free PMC article. Review.
-
FDA-Approved Inhibitors of RTK/Raf Signaling Potently Impair Multiple Steps of In Vitro and Ex Vivo Influenza A Virus Infections.Viruses. 2022 Sep 16;14(9):2058. doi: 10.3390/v14092058. Viruses. 2022. PMID: 36146864 Free PMC article.
-
Role of Virus-Induced EGFR Trafficking in Proviral Functions.Biomolecules. 2023 Dec 9;13(12):1766. doi: 10.3390/biom13121766. Biomolecules. 2023. PMID: 38136637 Free PMC article. Review.
-
Syk Facilitates Influenza A Virus Replication by Restraining Innate Immunity at the Late Stage of Viral Infection.J Virol. 2022 Apr 13;96(7):e0020022. doi: 10.1128/jvi.00200-22. Epub 2022 Mar 16. J Virol. 2022. PMID: 35293768 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
