

Amyotrophic Lateral Sclerosis: Proteins, Proteostasis, Prions, and Promises
- PMID: 33328890
- PMCID: PMC7671971
- DOI: 10.3389/fncel.2020.581907
Amyotrophic Lateral Sclerosis: Proteins, Proteostasis, Prions, and Promises
Abstract
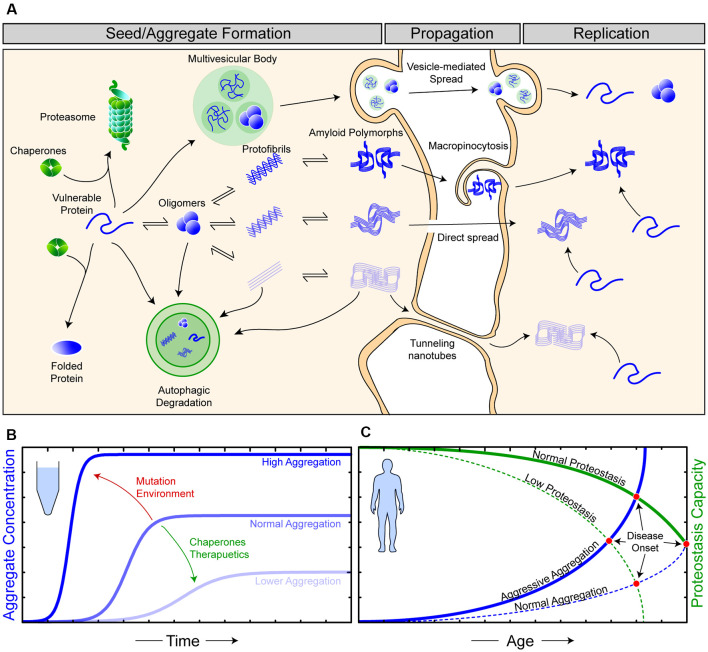
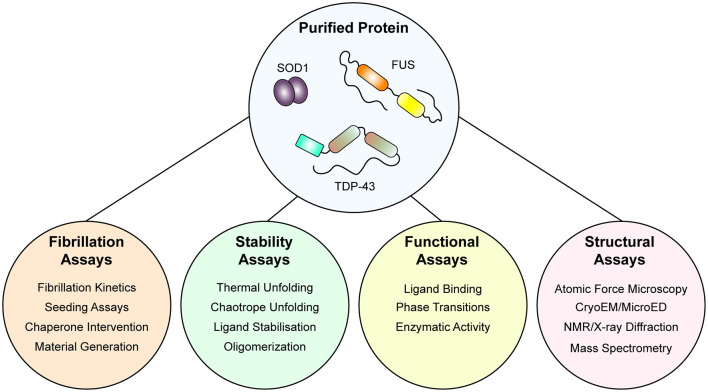
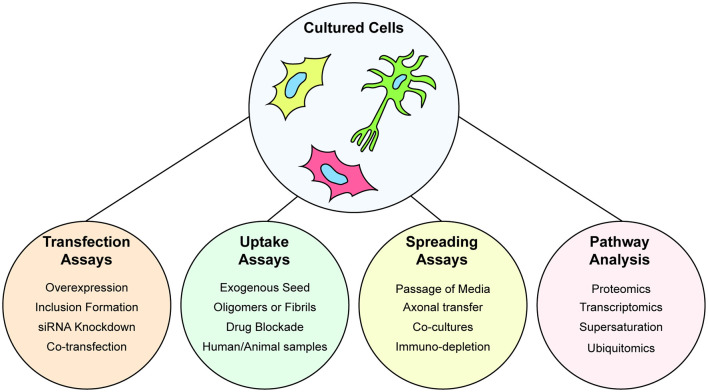
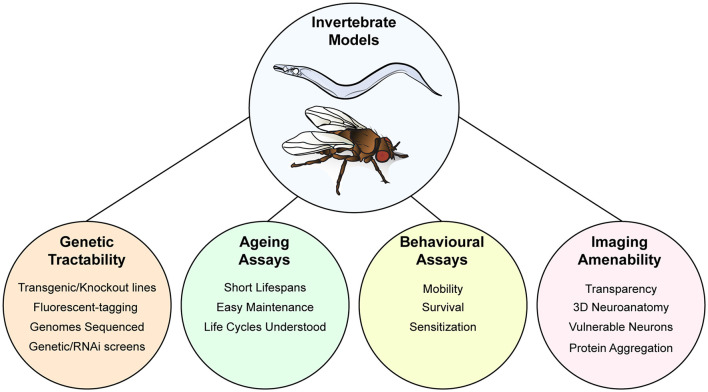
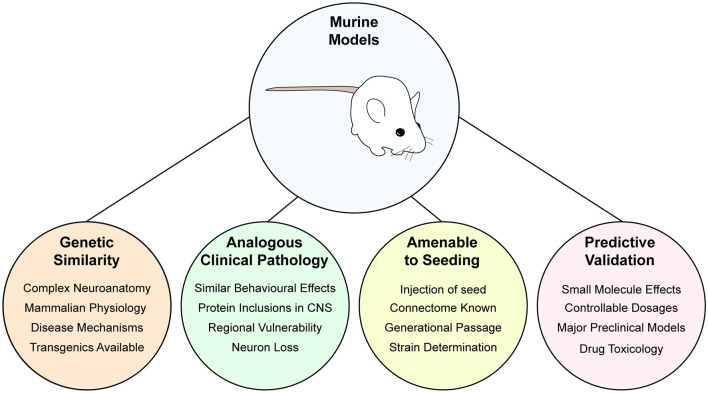
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of the motor neurons that innervate muscle, resulting in gradual paralysis and culminating in the inability to breathe or swallow. This neuronal degeneration occurs in a spatiotemporal manner from a point of onset in the central nervous system (CNS), suggesting that there is a molecule that spreads from cell-to-cell. There is strong evidence that the onset and progression of ALS pathology is a consequence of protein misfolding and aggregation. In line with this, a hallmark pathology of ALS is protein deposition and inclusion formation within motor neurons and surrounding glia of the proteins TAR DNA-binding protein 43, superoxide dismutase-1, or fused in sarcoma. Collectively, the observed protein aggregation, in conjunction with the spatiotemporal spread of symptoms, strongly suggests a prion-like propagation of protein aggregation occurs in ALS. In this review, we discuss the role of protein aggregation in ALS concerning protein homeostasis (proteostasis) mechanisms and prion-like propagation. Furthermore, we examine the experimental models used to investigate these processes, including in vitro assays, cultured cells, invertebrate models, and murine models. Finally, we evaluate the therapeutics that may best prevent the onset or spread of pathology in ALS and discuss what lies on the horizon for treating this currently incurable disease.
Keywords: amyotrophic lateral scelerosis; in vitro models; invertebrate models; mouse models; prion-like; protein aggregation; proteostasis; therapeutics.
Copyright © 2020 McAlary, Chew, Lum, Geraghty, Yerbury and Cashman.
Figures
Similar articles
-
The prion-like nature of amyotrophic lateral sclerosis.Prog Mol Biol Transl Sci. 2020;175:261-296. doi: 10.1016/bs.pmbts.2020.07.002. Epub 2020 Sep 1. Prog Mol Biol Transl Sci. 2020. PMID: 32958236 Review.
-
Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis.Front Mol Neurosci. 2019 Nov 1;12:262. doi: 10.3389/fnmol.2019.00262. eCollection 2019. Front Mol Neurosci. 2019. PMID: 31736708 Free PMC article.
-
From molecule to molecule and cell to cell: prion-like mechanisms in amyotrophic lateral sclerosis.Neurobiol Dis. 2015 May;77:257-65. doi: 10.1016/j.nbd.2015.02.009. Epub 2015 Feb 17. Neurobiol Dis. 2015. PMID: 25701498 Review.
-
[Prion-like Properties of Misfolded Cu/Zn-superoxide Dismutase in Amyotrophic Lateral Sclerosis: Update and Perspectives].Yakugaku Zasshi. 2019;139(7):1015-1019. doi: 10.1248/yakushi.18-00165-5. Yakugaku Zasshi. 2019. PMID: 31257248 Review. Japanese.
-
Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS).Neuropathol Appl Neurobiol. 2015 Aug;41(5):578-97. doi: 10.1111/nan.12206. Epub 2015 Apr 20. Neuropathol Appl Neurobiol. 2015. PMID: 25487060 Review.
Cited by
-
Taking Cellular Heterogeneity Into Consideration When Modeling Astrocyte Involvement in Amyotrophic Lateral Sclerosis Using Human Induced Pluripotent Stem Cells.Front Cell Neurosci. 2021 Sep 17;15:707861. doi: 10.3389/fncel.2021.707861. eCollection 2021. Front Cell Neurosci. 2021. PMID: 34602979 Free PMC article. Review.
-
Repetitive Mild Closed Head Injury in Adolescent Mice Is Associated with Impaired Proteostasis, Neuroinflammation, and Tauopathy.J Neurosci. 2022 Mar 23;42(12):2418-2432. doi: 10.1523/JNEUROSCI.0682-21.2021. Epub 2022 Feb 1. J Neurosci. 2022. PMID: 35105673 Free PMC article.
-
Rapid Generation of Ventral Spinal Cord-like Astrocytes from Human iPSCs for Modeling Non-Cell Autonomous Mechanisms of Lower Motor Neuron Disease.Cells. 2022 Jan 24;11(3):399. doi: 10.3390/cells11030399. Cells. 2022. PMID: 35159209 Free PMC article.
-
Matrin3: Disorder and ALS Pathogenesis.Front Mol Biosci. 2022 Jan 10;8:794646. doi: 10.3389/fmolb.2021.794646. eCollection 2021. Front Mol Biosci. 2022. PMID: 35083279 Free PMC article. Review.
-
Protein nanocondensates: the next frontier.Biophys Rev. 2023 Aug 9;15(4):515-530. doi: 10.1007/s12551-023-01105-1. eCollection 2023 Aug. Biophys Rev. 2023. PMID: 37681092 Free PMC article. Review.
References
-
- Abdolvahabi A., Shi Y., Rasouli S., Croom C. M., Aliyan A., Martí A. A., et al. . (2017). Kaplan-meier meets chemical kinetics: intrinsic rate of SOD1 amyloidogenesis decreased by subset of als mutations and cannot fully explain age of disease onset. ACS Chem. Neurosci. 8, 1378–1389. 10.1021/acschemneuro.7b00029 - DOI - PubMed
-
- Abe K., Itoyama Y., Sobue G., Tsuji S., Aoki M., Doyu M., et al. . (2014). Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 610–617. 10.3109/21678421.2014.959024 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous