

Targeting transcriptional coregulator OCA-B/Pou2af1 blocks activated autoreactive T cells in the pancreas and type 1 diabetes
- PMID: 33295943
- PMCID: PMC7731945
- DOI: 10.1084/jem.20200533
Targeting transcriptional coregulator OCA-B/Pou2af1 blocks activated autoreactive T cells in the pancreas and type 1 diabetes
Abstract
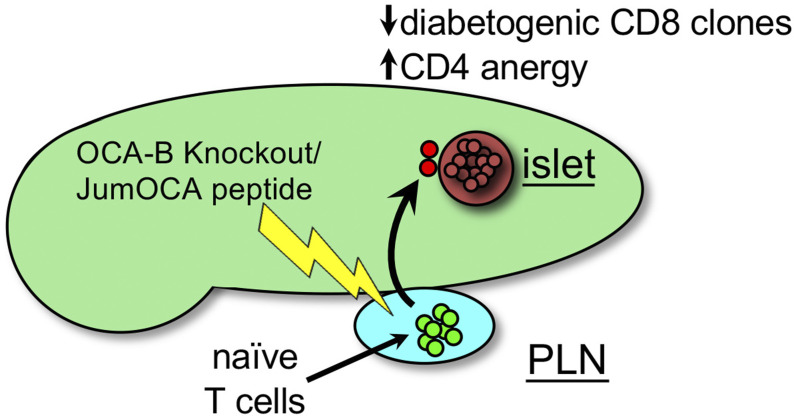
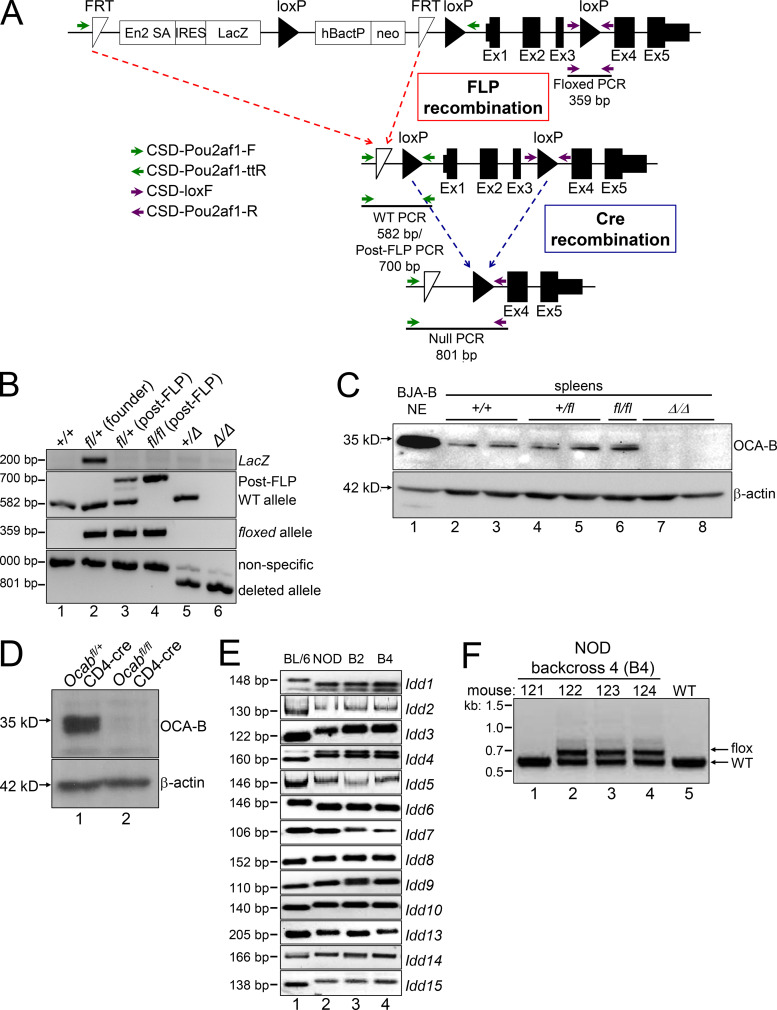
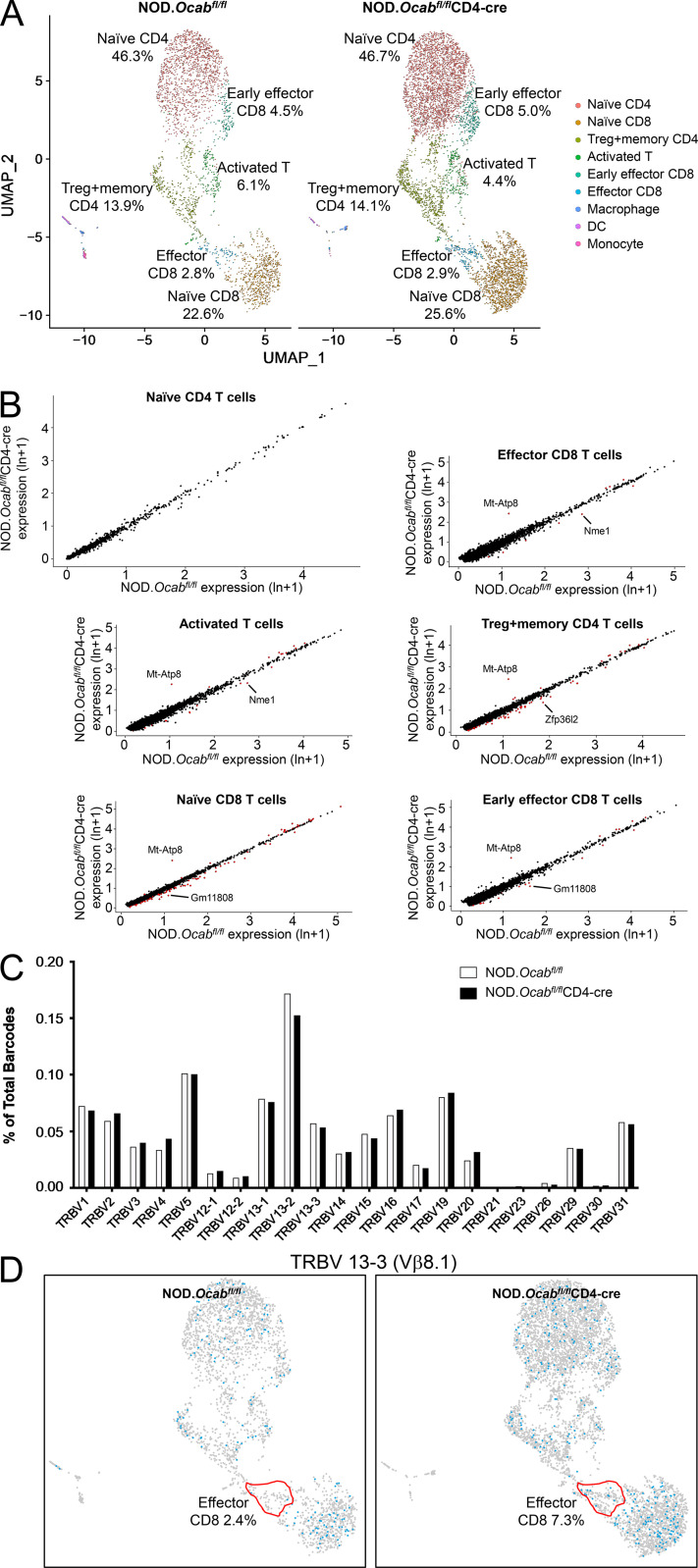
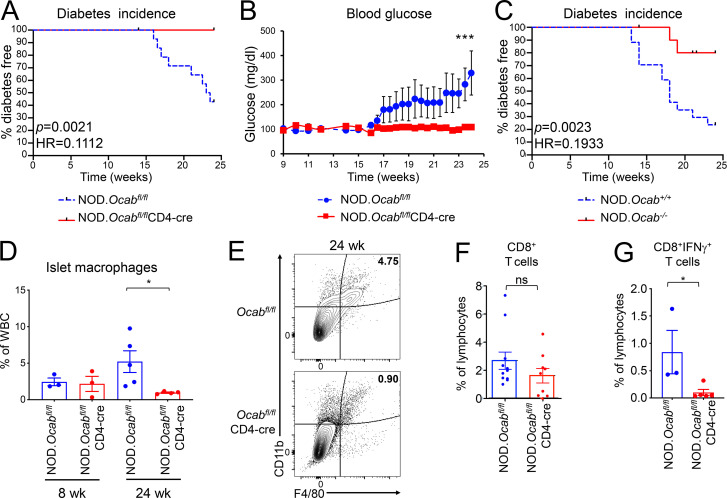
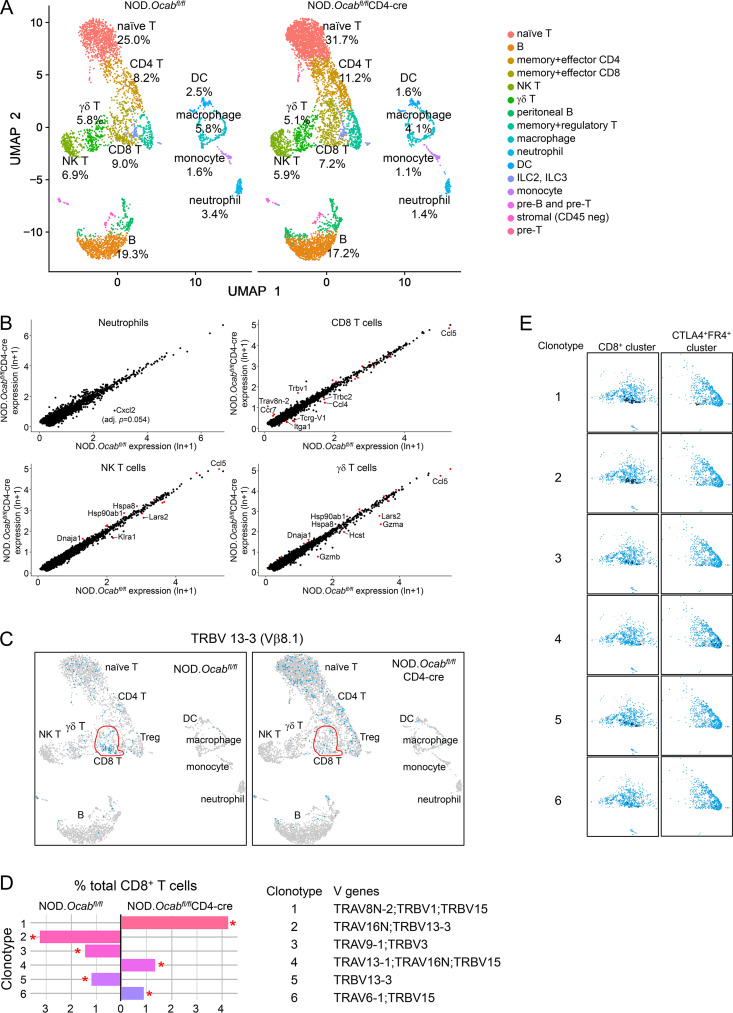
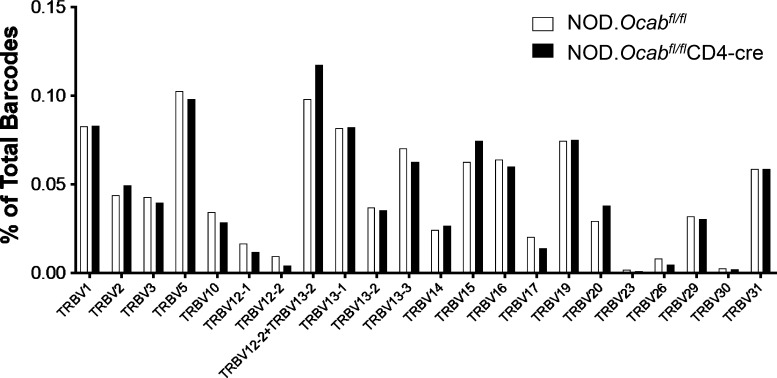
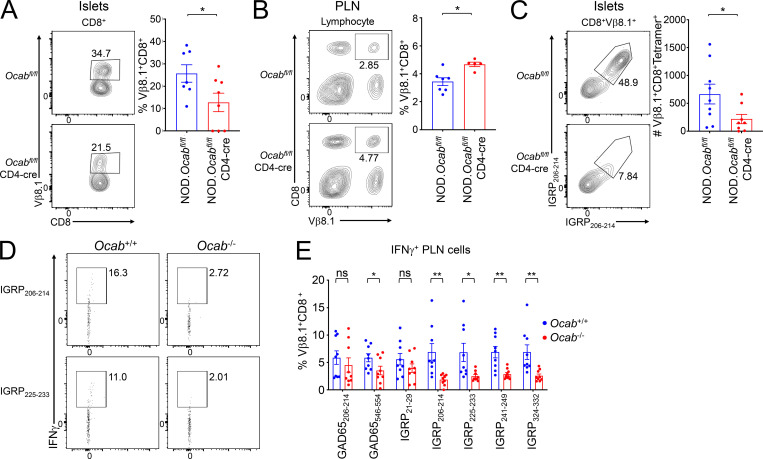
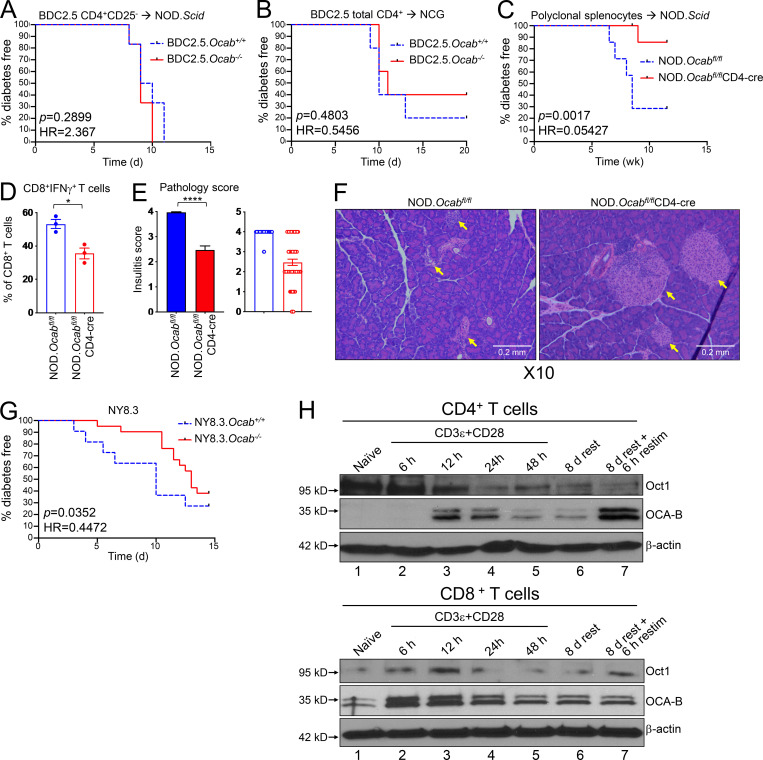
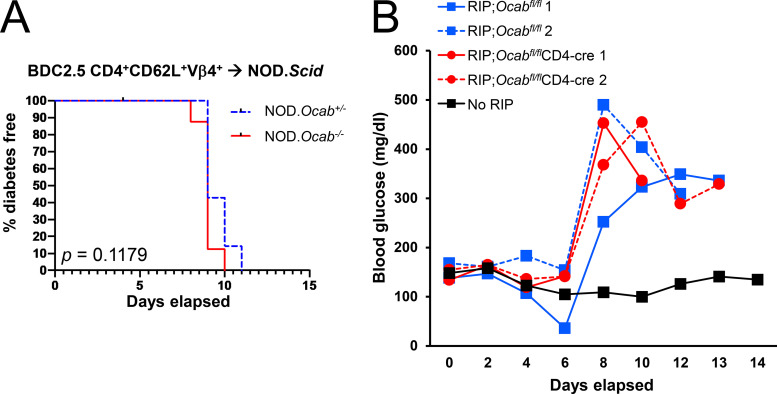
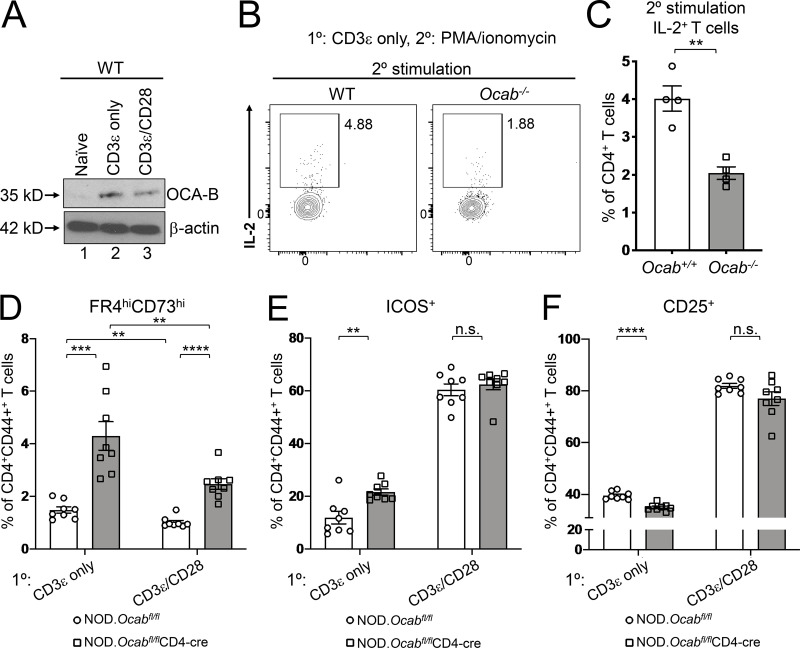
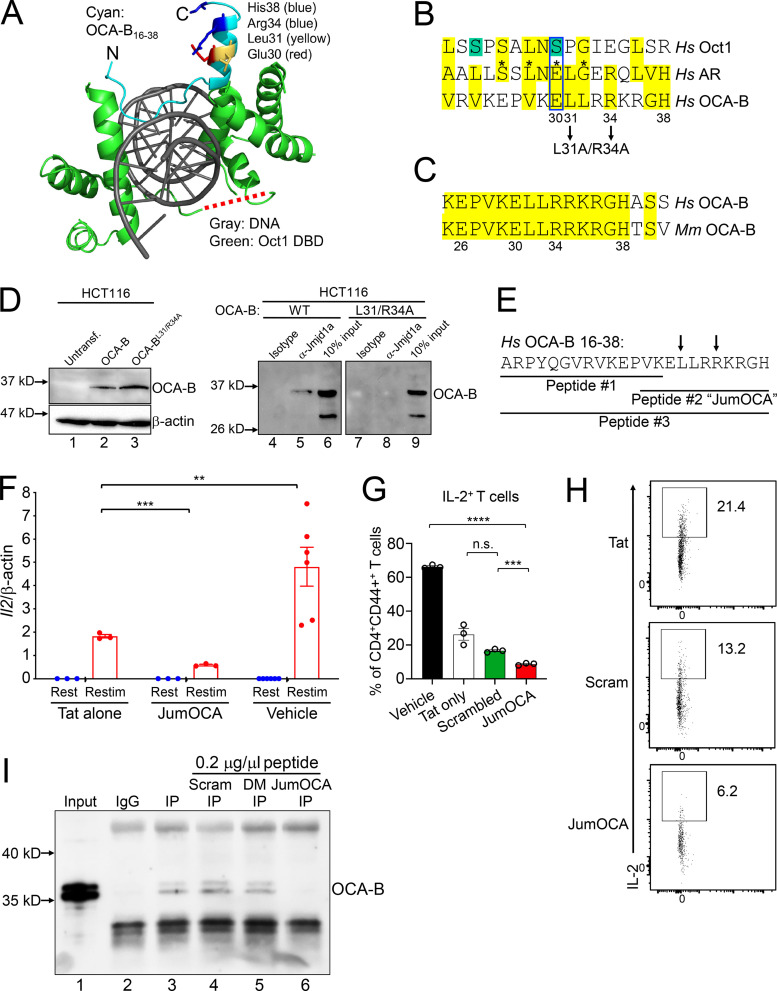
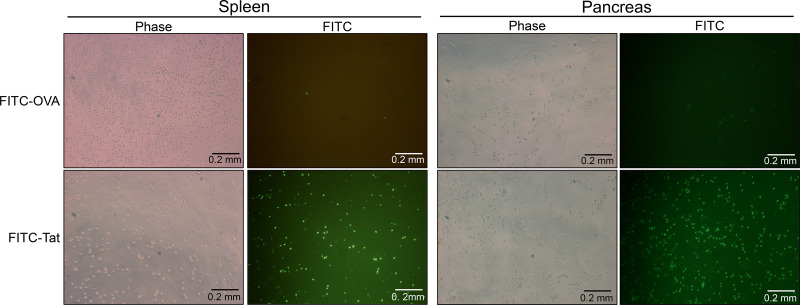
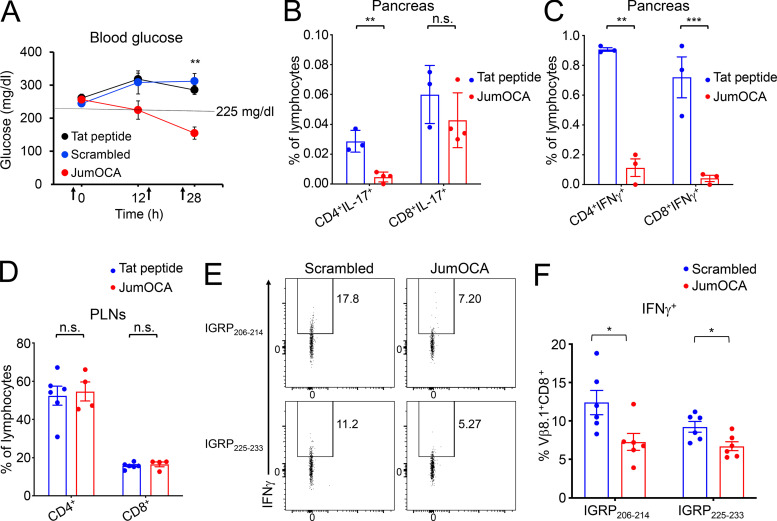
The transcriptional coregulator OCA-B promotes expression of T cell target genes in cases of repeated antigen exposure, a necessary feature of autoimmunity. We hypothesized that T cell-specific OCA-B deletion and pharmacologic OCA-B inhibition would protect mice from autoimmune diabetes. We developed an Ocab conditional allele and backcrossed it onto a diabetes-prone NOD/ShiLtJ strain background. T cell-specific OCA-B loss protected mice from spontaneous disease. Protection was associated with large reductions in islet CD8+ T cell receptor specificities associated with diabetes pathogenesis. CD4+ clones associated with diabetes were present but associated with anergic phenotypes. The protective effect of OCA-B loss was recapitulated using autoantigen-specific NY8.3 mice but diminished in monoclonal models specific to artificial or neoantigens. Rationally designed membrane-penetrating OCA-B peptide inhibitors normalized glucose levels and reduced T cell infiltration and proinflammatory cytokine expression in newly diabetic NOD mice. Together, the results indicate that OCA-B is a potent autoimmune regulator and a promising target for pharmacologic inhibition.
© 2020 Kim et al.
Conflict of interest statement
Disclosures: D. Tantin reported a patent (62/666,325). No other disclosures were reported.
Figures
Similar articles
-
Genetic and therapeutic control of diabetogenic CD8+ T cells.Novartis Found Symp. 2008;292:130-6; discussion 136-45, 202-3. doi: 10.1002/9780470697405.ch12. Novartis Found Symp. 2008. PMID: 19209463
-
Pancreatic infiltration but not diabetes occurs in the relative absence of MHC class II-restricted CD4 T cells: studies using NOD/CIITA-deficient mice.J Immunol. 1999 Apr 15;162(8):4576-88. J Immunol. 1999. PMID: 10201997
-
Rotavirus acceleration of murine type 1 diabetes is associated with increased MHC class I-restricted antigen presentation by B cells and elevated proinflammatory cytokine expression by T cells.Virus Res. 2014 Jan 22;179:73-84. doi: 10.1016/j.virusres.2013.11.009. Epub 2013 Nov 15. Virus Res. 2014. PMID: 24246305
-
T-cell epitopes in type 1 diabetes.Curr Diab Rep. 2004 Apr;4(2):87-94. doi: 10.1007/s11892-004-0062-0. Curr Diab Rep. 2004. PMID: 15035967 Review.
-
Autoimmune diabetes: the role of T cells, MHC molecules and autoantigens.Autoimmunity. 1998;27(3):159-77. doi: 10.3109/08916939809003864. Autoimmunity. 1998. PMID: 9609134 Review.
Cited by
-
WDFY4 deficiency in NOD mice ameliorates autoimmune diabetes and insulitis.Proc Natl Acad Sci U S A. 2023 Mar 28;120(13):e2219956120. doi: 10.1073/pnas.2219956120. Epub 2023 Mar 20. Proc Natl Acad Sci U S A. 2023. PMID: 36940342 Free PMC article.
-
Construction and Validation of a Tumor Microenvironment-Based Scoring System to Evaluate Prognosis and Response to Immune Checkpoint Inhibitor Therapy in Lung Adenocarcinoma Patients.Genes (Basel). 2022 May 26;13(6):951. doi: 10.3390/genes13060951. Genes (Basel). 2022. PMID: 35741714 Free PMC article.
-
Deficiency of Trex1 leads to spontaneous development of type 1 diabetes.Nutr Metab (Lond). 2024 Jan 2;21(1):2. doi: 10.1186/s12986-023-00777-6. Nutr Metab (Lond). 2024. PMID: 38166933 Free PMC article.
-
A novel iTreg-related signature for prognostic prediction in lung adenocarcinoma.Cancer Sci. 2024 Jan;115(1):109-124. doi: 10.1111/cas.16015. Epub 2023 Nov 27. Cancer Sci. 2024. PMID: 38015097 Free PMC article.
-
Pancreatic draining lymph nodes (PLNs) serve as a pathogenic hub contributing to the development of type 1 diabetes.Cell Biosci. 2023 Aug 28;13(1):156. doi: 10.1186/s13578-023-01110-7. Cell Biosci. 2023. PMID: 37641145 Free PMC article. Review.
References
-
- Ban, M., Booth D., Heard R., Stewart G., Goris A., Vandenbroeck K., Dubois B., Laaksonen M., Ilonen J., Alizadeh M., et al. . Games Collaborative Group . 2006. Linkage disequilibrium screening for multiple sclerosis implicates JAG1 and POU2AF1 as susceptibility genes in Europeans. J. Neuroimmunol. 179:108–116. 10.1016/j.jneuroim.2006.06.003 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
