

Podocyte EGFR Inhibits Autophagy Through Upregulation of Rubicon in Type 2 Diabetic Nephropathy
- PMID: 33239448
- PMCID: PMC7881855
- DOI: 10.2337/db20-0660
Podocyte EGFR Inhibits Autophagy Through Upregulation of Rubicon in Type 2 Diabetic Nephropathy
Abstract
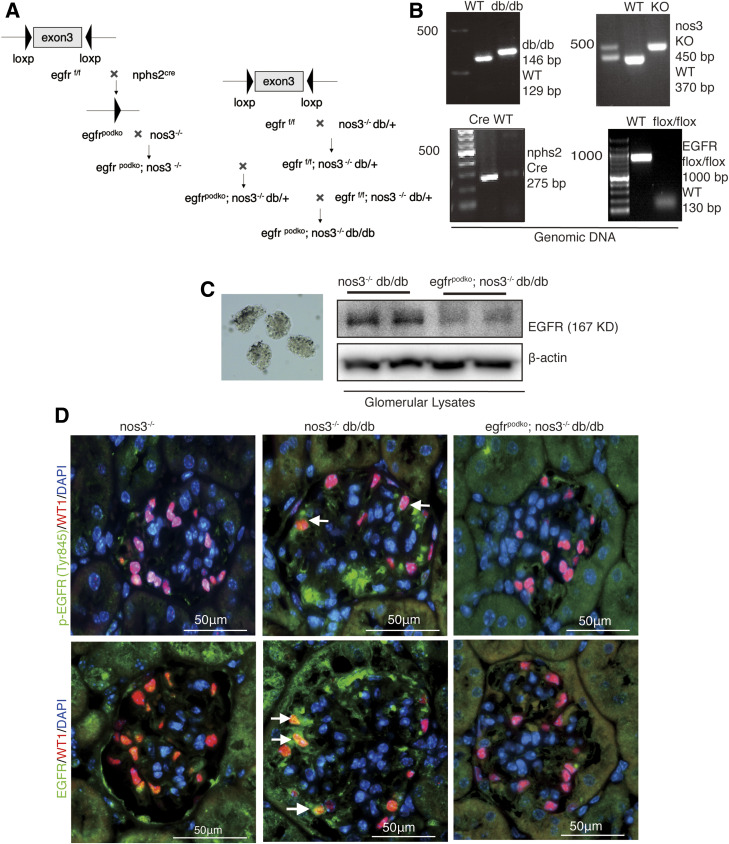
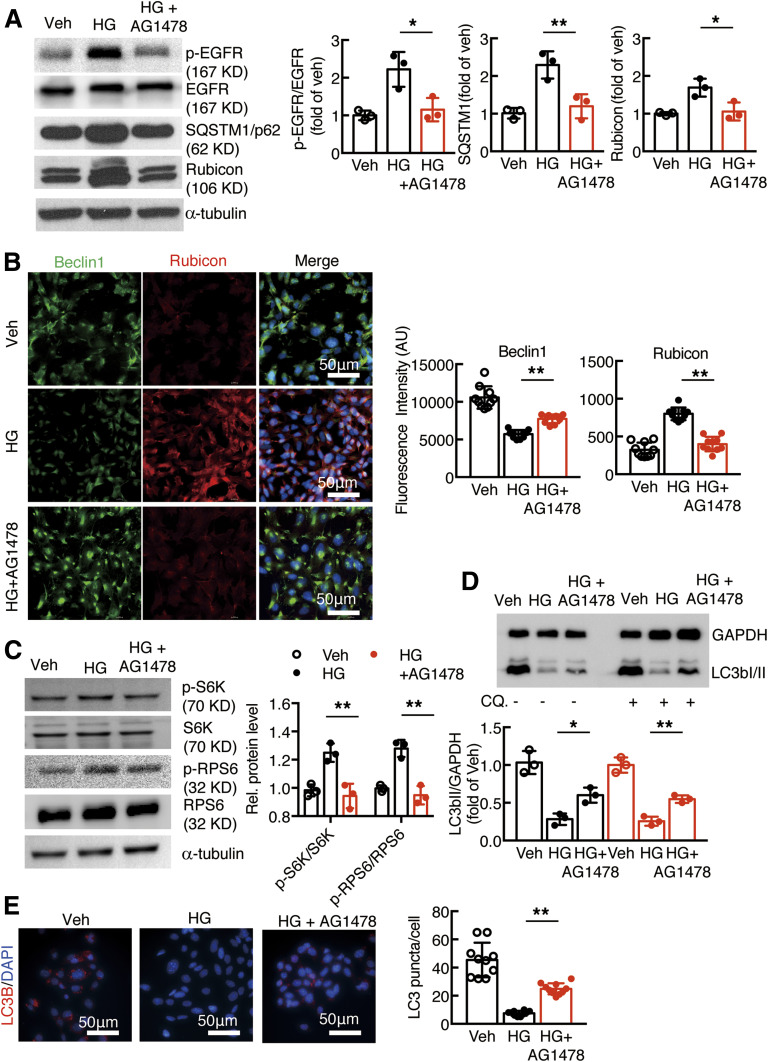
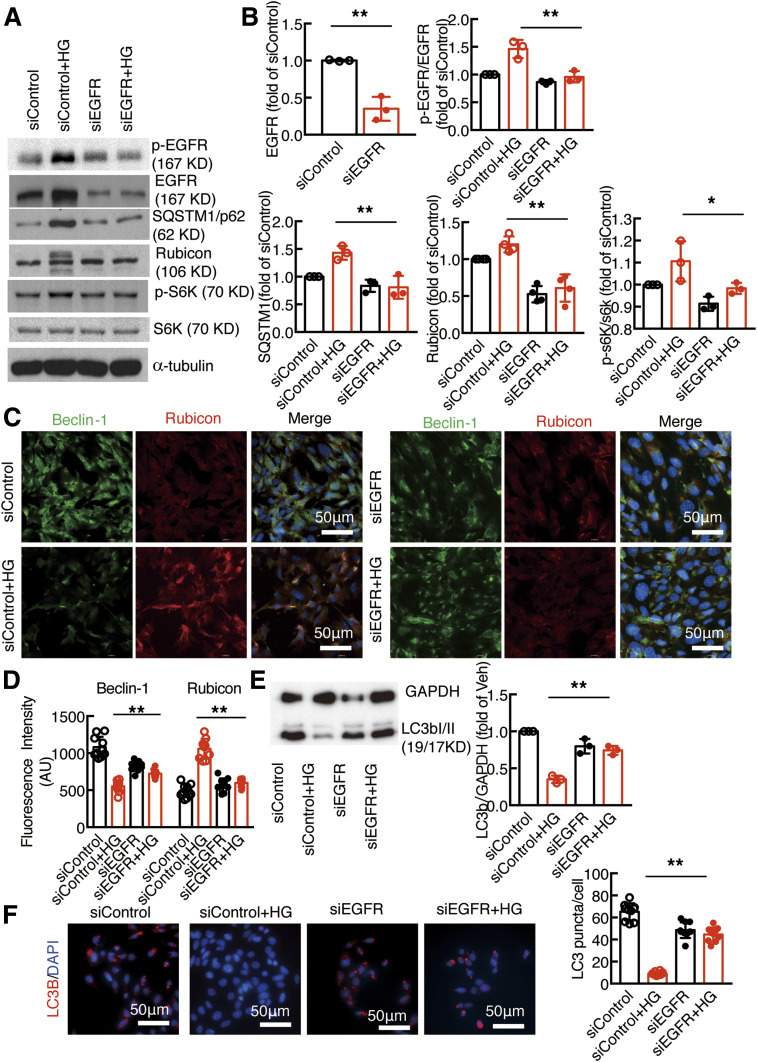
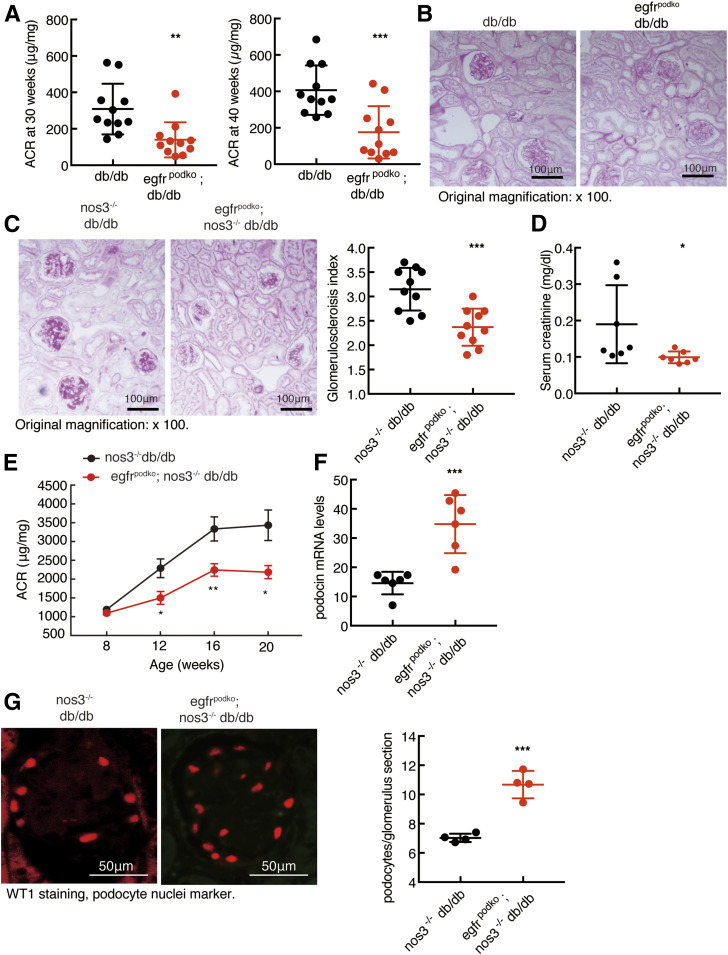
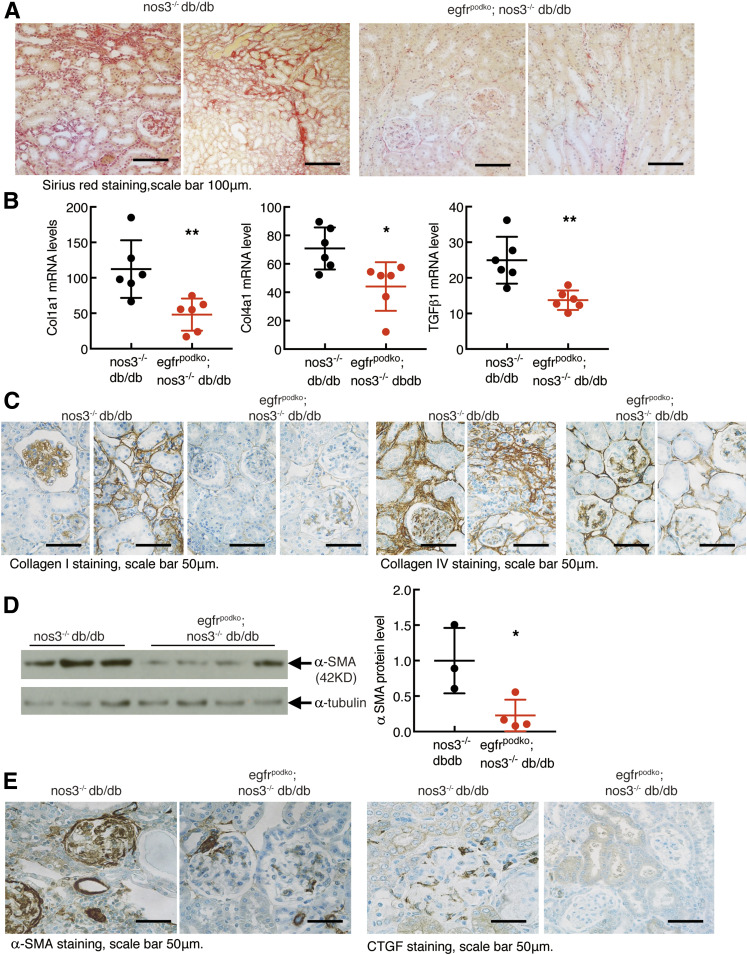
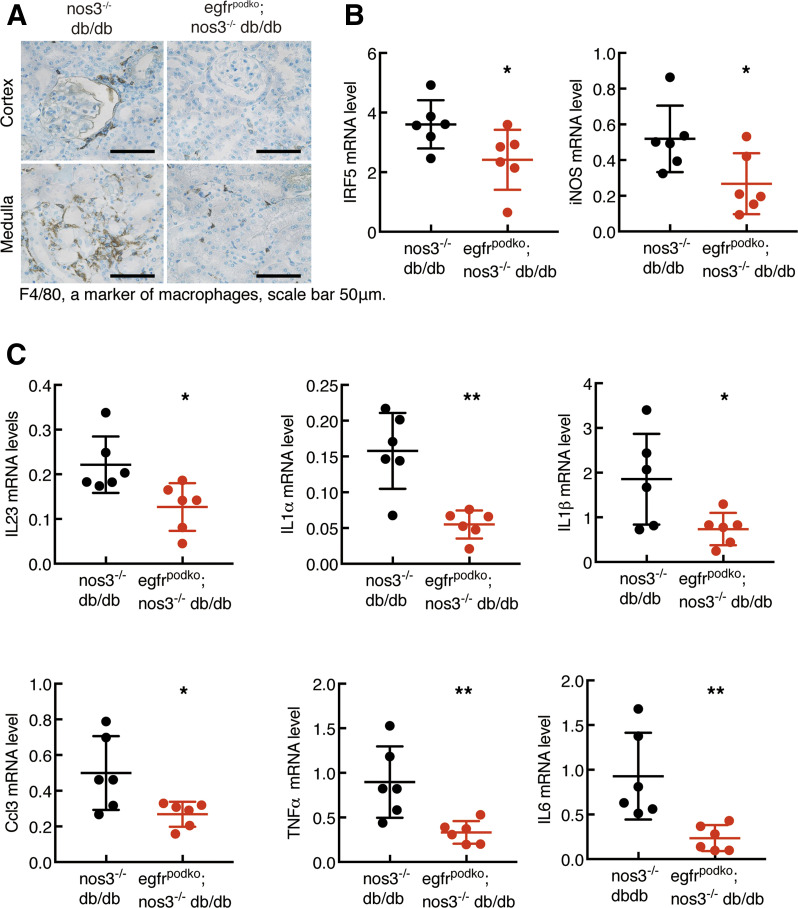
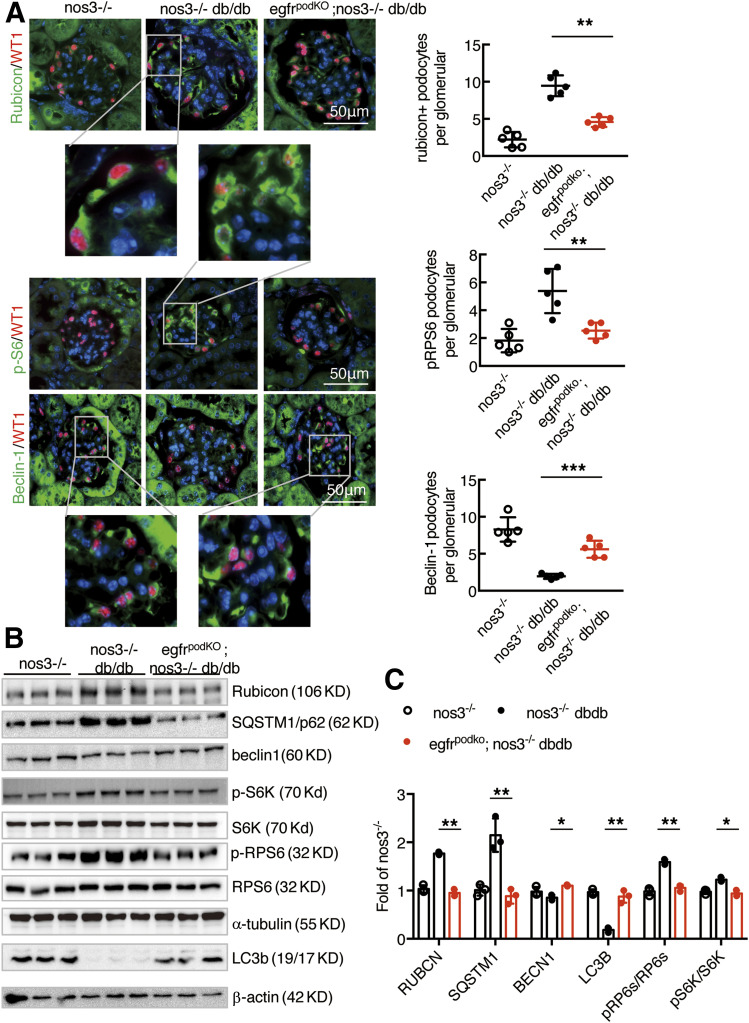
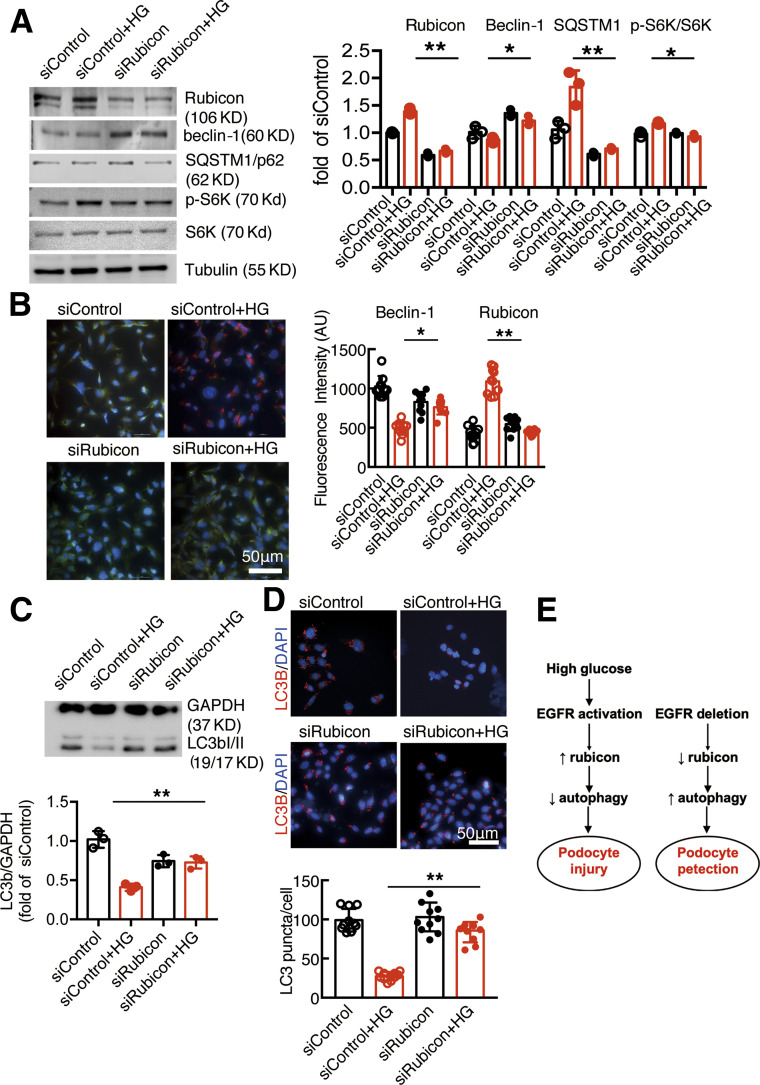
Renal epidermal growth factor receptor (EGFR) signaling is activated in models of diabetic nephropathy (DN), and inhibition of the EGFR signaling pathway protects against the development of DN. We have now determined that in cultured podocytes, high glucose led to increases in activation of EGFR signaling but decreases in autophagy activity as indicated by decreased beclin-1 and inhibition of LC3B autophagosome formation as well as increased rubicon (an autophagy inhibitor) and SQSTM1 (autophagy substrate). Either genetic (small interfering [si]EGFR) or pharmacologic (AG1478) inhibition of EGFR signaling attenuated the decreased autophagy activity. In addition, rubicon siRNA knockdown prevented high glucose-induced inhibition of autophagy in podocytes. We further examined whether selective EGFR deletion in podocytes affected the progression of DN in type 2 diabetes. Selective podocyte EGFR deletion had no effect on body weight or fasting blood sugars in either db/db mice or nos3 -/-; db/db mice, a model of accelerated type 2 DN. However selective podocyte EGFR deletion led to relative podocyte preservation and marked reduction in albuminuria and glomerulosclerosis, renal proinflammatory cytokine/chemokine expression, and decreased profibrotic and fibrotic components in nos3 -/-; db/db mice. Podocyte EGFR deletion led to decreased podocyte expression of rubicon, in association with increased podocyte autophagy activity. Therefore, activation of EGFR signaling in podocytes contributes to progression of DN at least in part by increasing rubicon expression, leading to subsequent autophagy inhibition and podocyte injury.
© 2020 by the American Diabetes Association.
Figures
Similar articles
-
Inhibition of Epidermal Growth Factor Receptor Activation Is Associated With Improved Diabetic Nephropathy and Insulin Resistance in Type 2 Diabetes.Diabetes. 2018 Sep;67(9):1847-1857. doi: 10.2337/db17-1513. Epub 2018 Jun 29. Diabetes. 2018. PMID: 29959129 Free PMC article.
-
NFAT2 inhibitor ameliorates diabetic nephropathy and podocyte injury in db/db mice.Br J Pharmacol. 2013 Sep;170(2):426-39. doi: 10.1111/bph.12292. Br J Pharmacol. 2013. PMID: 23826864 Free PMC article.
-
EGF receptor deletion in podocytes attenuates diabetic nephropathy.J Am Soc Nephrol. 2015 May;26(5):1115-25. doi: 10.1681/ASN.2014020192. Epub 2014 Sep 3. J Am Soc Nephrol. 2015. PMID: 25185988 Free PMC article.
-
Research Progress on the Pathological Mechanisms of Podocytes in Diabetic Nephropathy.J Diabetes Res. 2020 Jul 8;2020:7504798. doi: 10.1155/2020/7504798. eCollection 2020. J Diabetes Res. 2020. PMID: 32695831 Free PMC article. Review.
-
Nutrient sensing, signaling transduction, and autophagy in podocyte injury: implications for kidney disease.J Nephrol. 2023 Jan;36(1):17-29. doi: 10.1007/s40620-022-01365-2. Epub 2022 Jun 15. J Nephrol. 2023. PMID: 35704261 Review.
Cited by
-
Circ-0000953 deficiency exacerbates podocyte injury and autophagy disorder by targeting Mir665-3p-Atg4b in diabetic nephropathy.Autophagy. 2024 May;20(5):1072-1097. doi: 10.1080/15548627.2023.2286128. Epub 2023 Dec 5. Autophagy. 2024. PMID: 38050963 Free PMC article.
-
"Qi Nan" agarwood restores podocyte autophagy in diabetic kidney disease by targeting EGFR signaling pathway.Chin Med. 2024 Apr 23;19(1):63. doi: 10.1186/s13020-024-00923-z. Chin Med. 2024. PMID: 38654354 Free PMC article.
-
Targeting the epidermal growth factor receptor (EGFR/ErbB) for the potential treatment of renal pathologies.Front Pharmacol. 2024 Aug 21;15:1394997. doi: 10.3389/fphar.2024.1394997. eCollection 2024. Front Pharmacol. 2024. PMID: 39234105 Free PMC article. Review.
-
Single-Cell RNA Sequencing Profiles Identify Important Pathophysiologic Factors in the Progression of Diabetic Nephropathy.Front Cell Dev Biol. 2022 May 10;10:798316. doi: 10.3389/fcell.2022.798316. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35620059 Free PMC article.
-
Uncovering the mechanism of Qidan Dihuang Granule in the treatment of diabetic kidney disease combined network pharmacology, UHPLC-MS/MS with experimental validation.Heliyon. 2023 Oct 28;9(11):e21714. doi: 10.1016/j.heliyon.2023.e21714. eCollection 2023 Nov. Heliyon. 2023. PMID: 37954274 Free PMC article.
References
-
- Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol 2013;9:713–723 - PubMed
-
- Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 2005;54:1626–1634 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
