

Structural and Functional Diversity of Resistance-Nodulation-Cell Division Transporters
- PMID: 33211490
- PMCID: PMC8119314
- DOI: 10.1021/acs.chemrev.0c00621
Structural and Functional Diversity of Resistance-Nodulation-Cell Division Transporters
Abstract
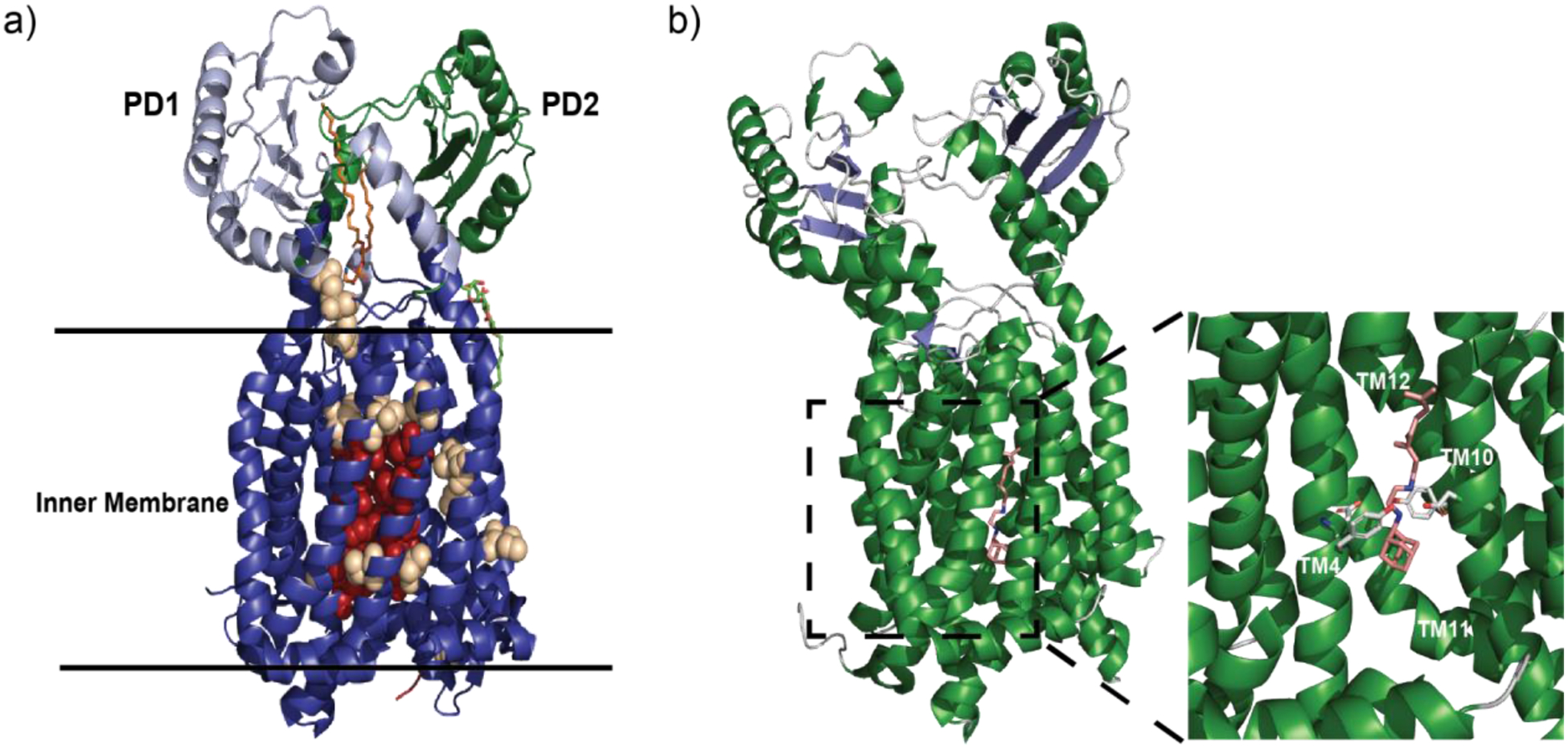
Multidrug resistant (MDR) bacteria are a global threat with many common infections becoming increasingly difficult to eliminate. While significant effort has gone into the development of potent biocides, the effectiveness of many first-line antibiotics has been diminished due to adaptive resistance mechanisms. Bacterial membrane proteins belonging to the resistance-nodulation-cell division (RND) superfamily play significant roles in mediating bacterial resistance to antimicrobials. They participate in multidrug efflux and cell wall biogenesis to transform bacterial pathogens into "superbugs" that are resistant even to last resort antibiotics. In this review, we summarize the RND superfamily of efflux transporters with a primary focus on the assembly and function of the inner membrane pumps. These pumps are critical for extrusion of antibiotics from the cell as well as the transport of lipid moieties to the outer membrane to establish membrane rigidity and stability. We analyze recently solved structures of bacterial inner membrane efflux pumps as to how they bind and transport their substrates. Our cumulative data indicate that these RND membrane proteins are able to utilize different oligomerization states to achieve particular activities, including forming MDR pumps and cell wall remodeling machineries, to ensure bacterial survival. This mechanistic insight, combined with simulated docking techniques, allows for the design and optimization of new efflux pump inhibitors to more effectively treat infections that today are difficult or impossible to cure.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

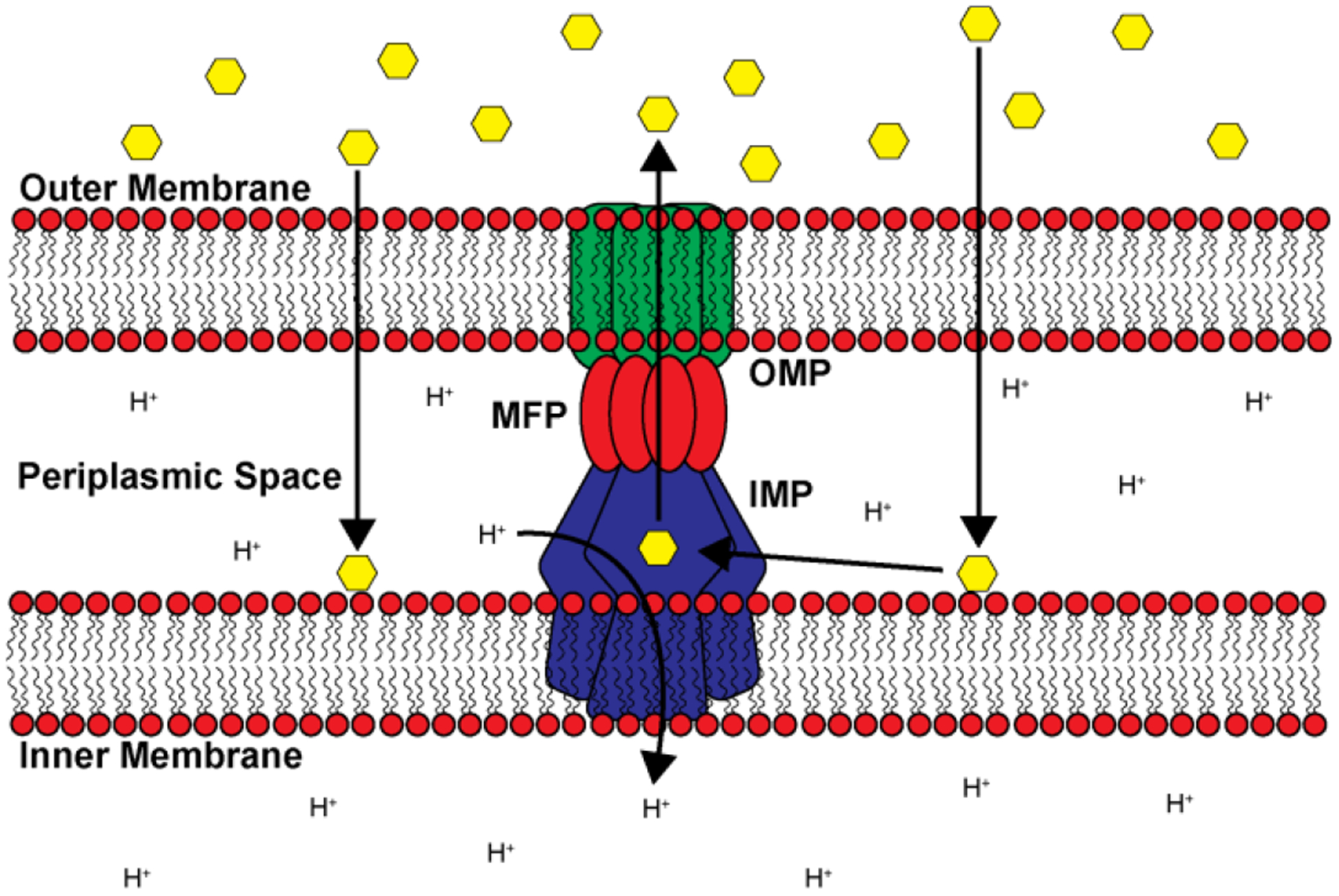

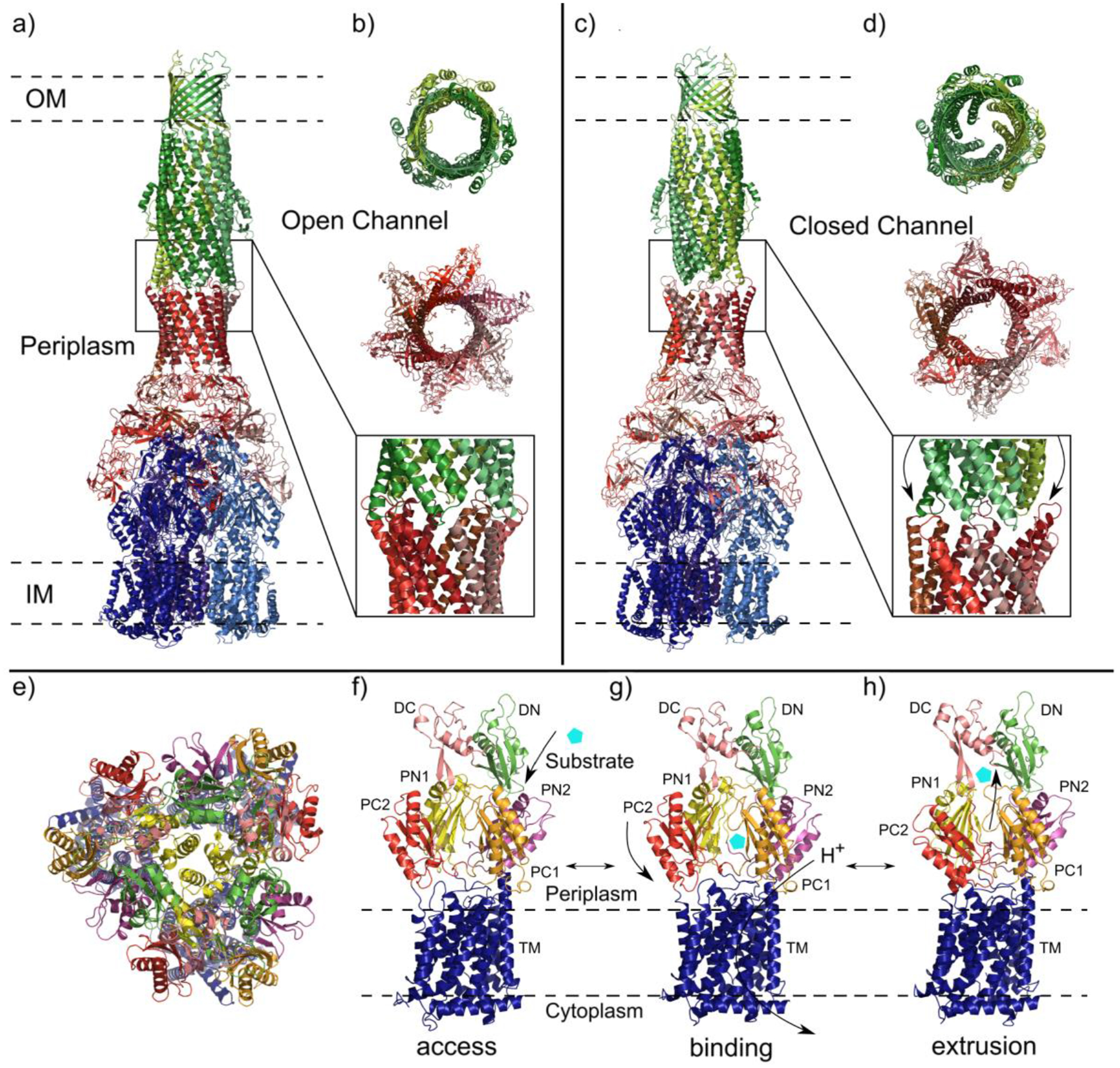
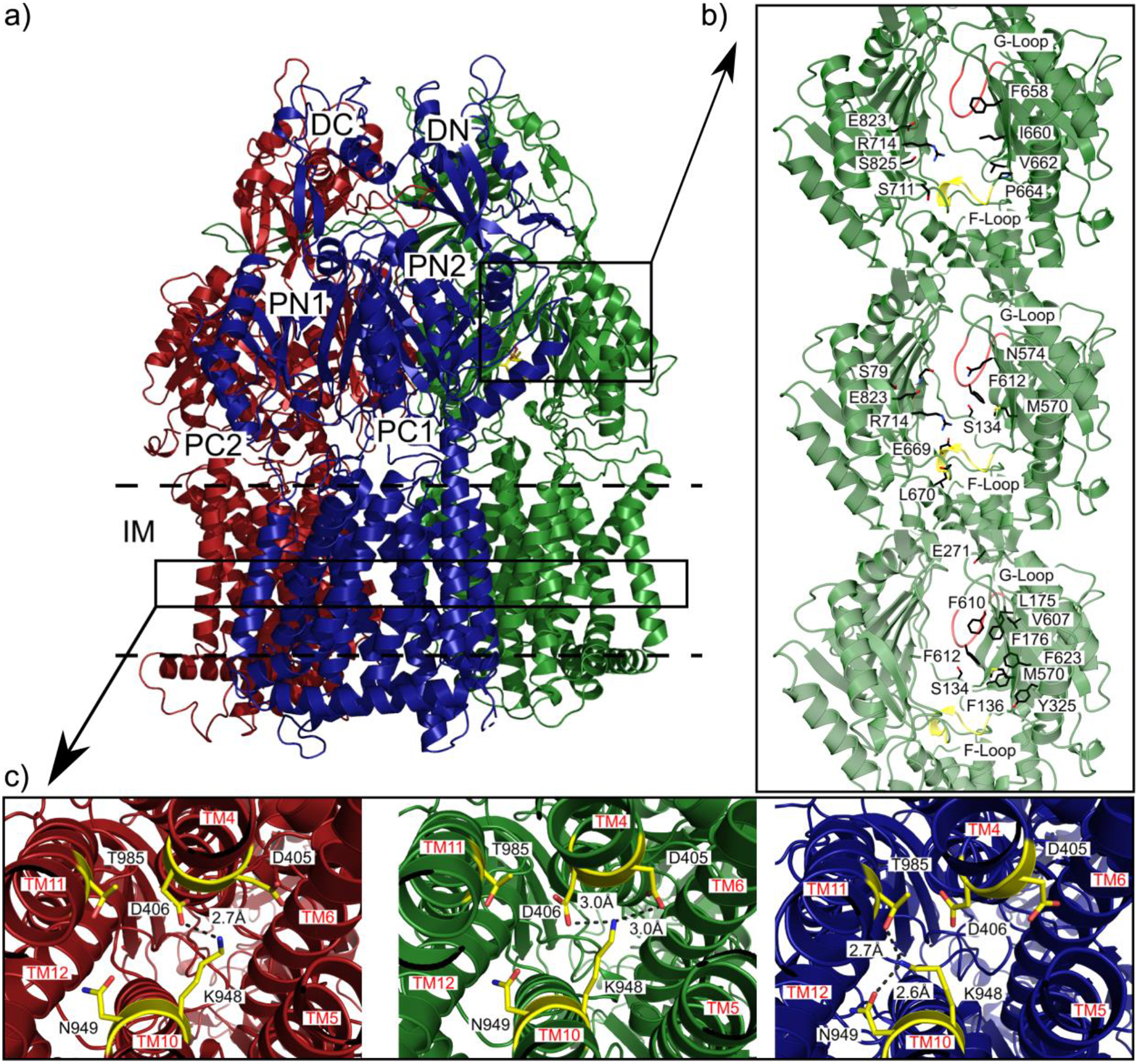
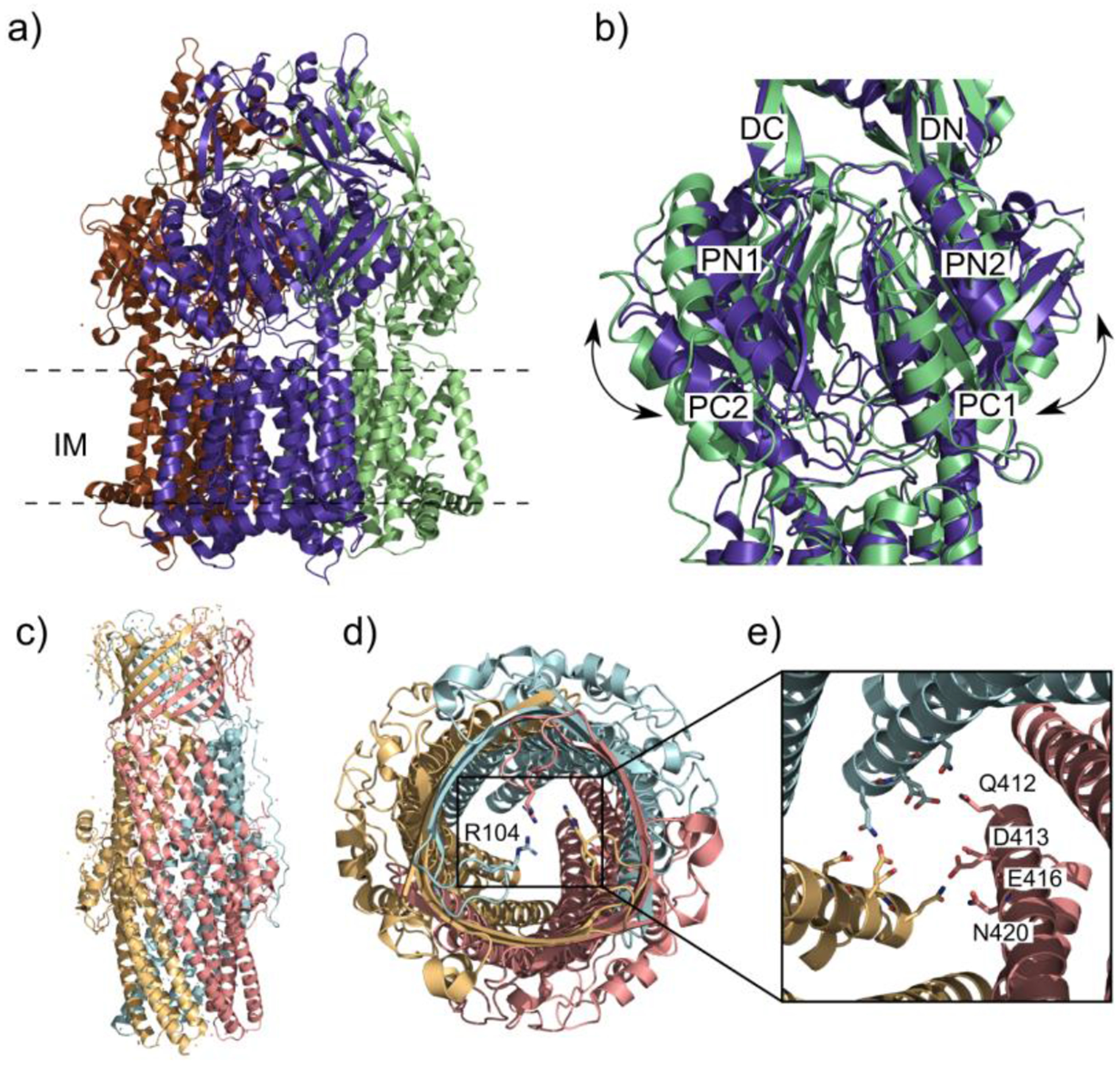
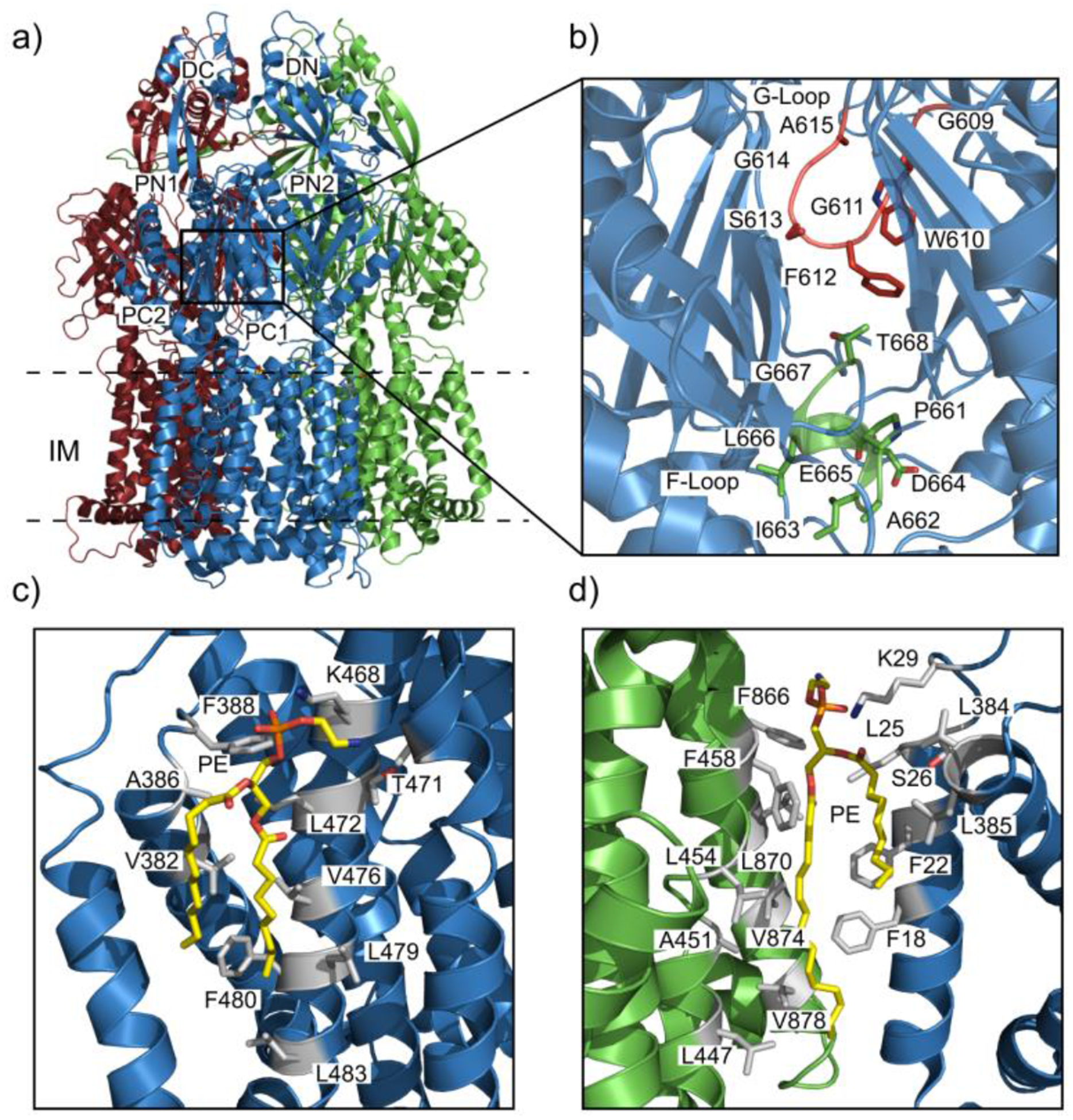
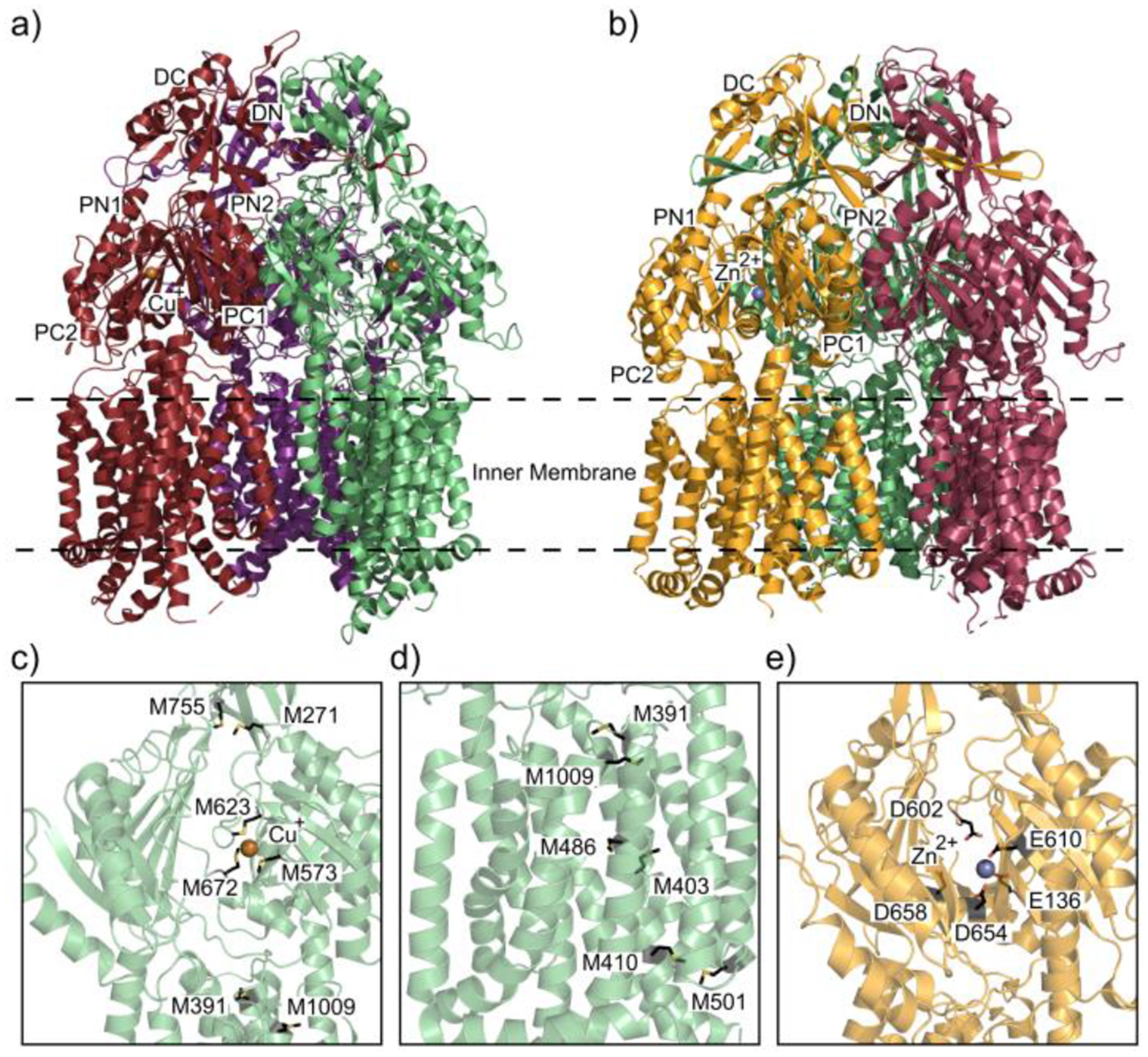
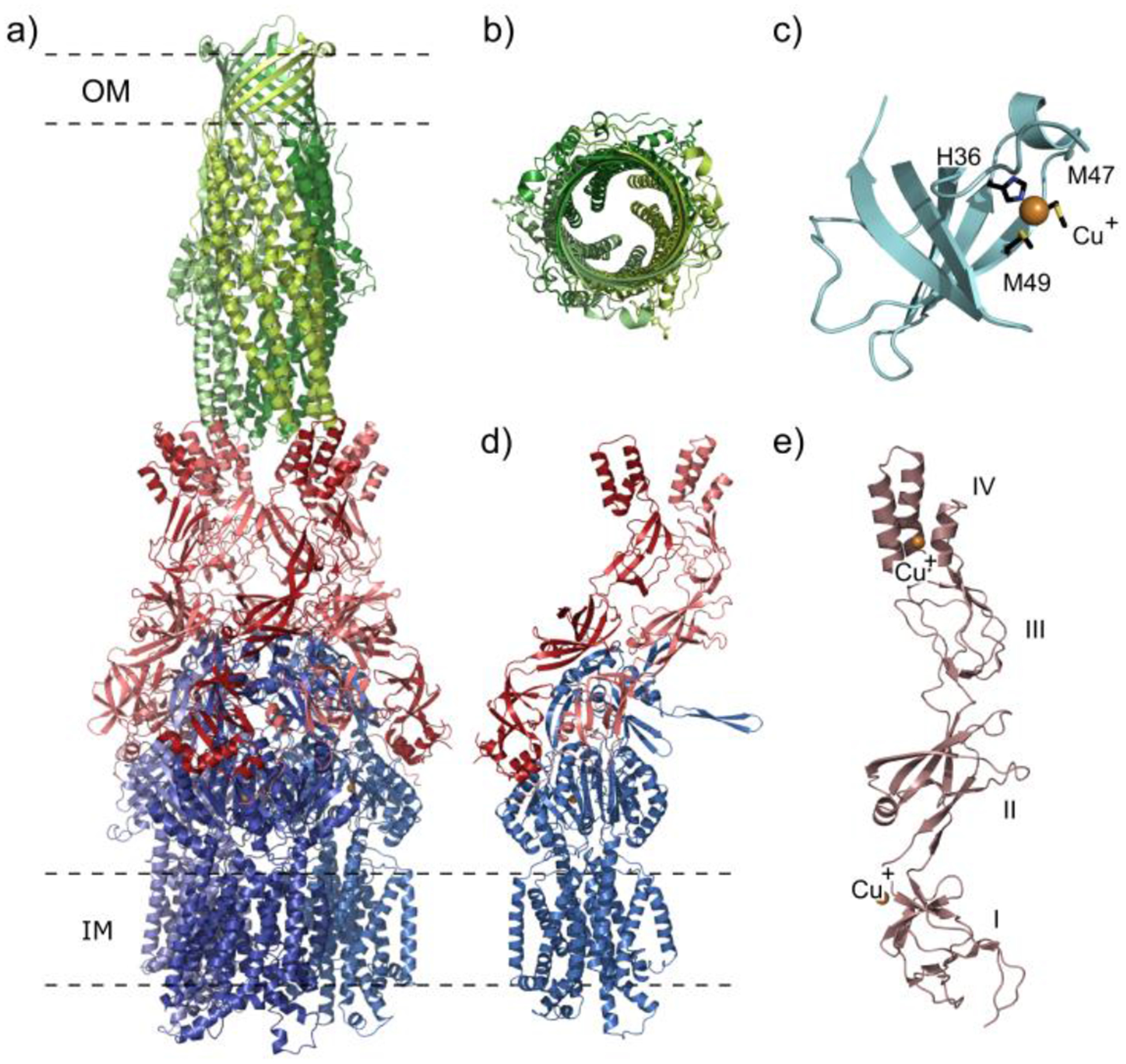
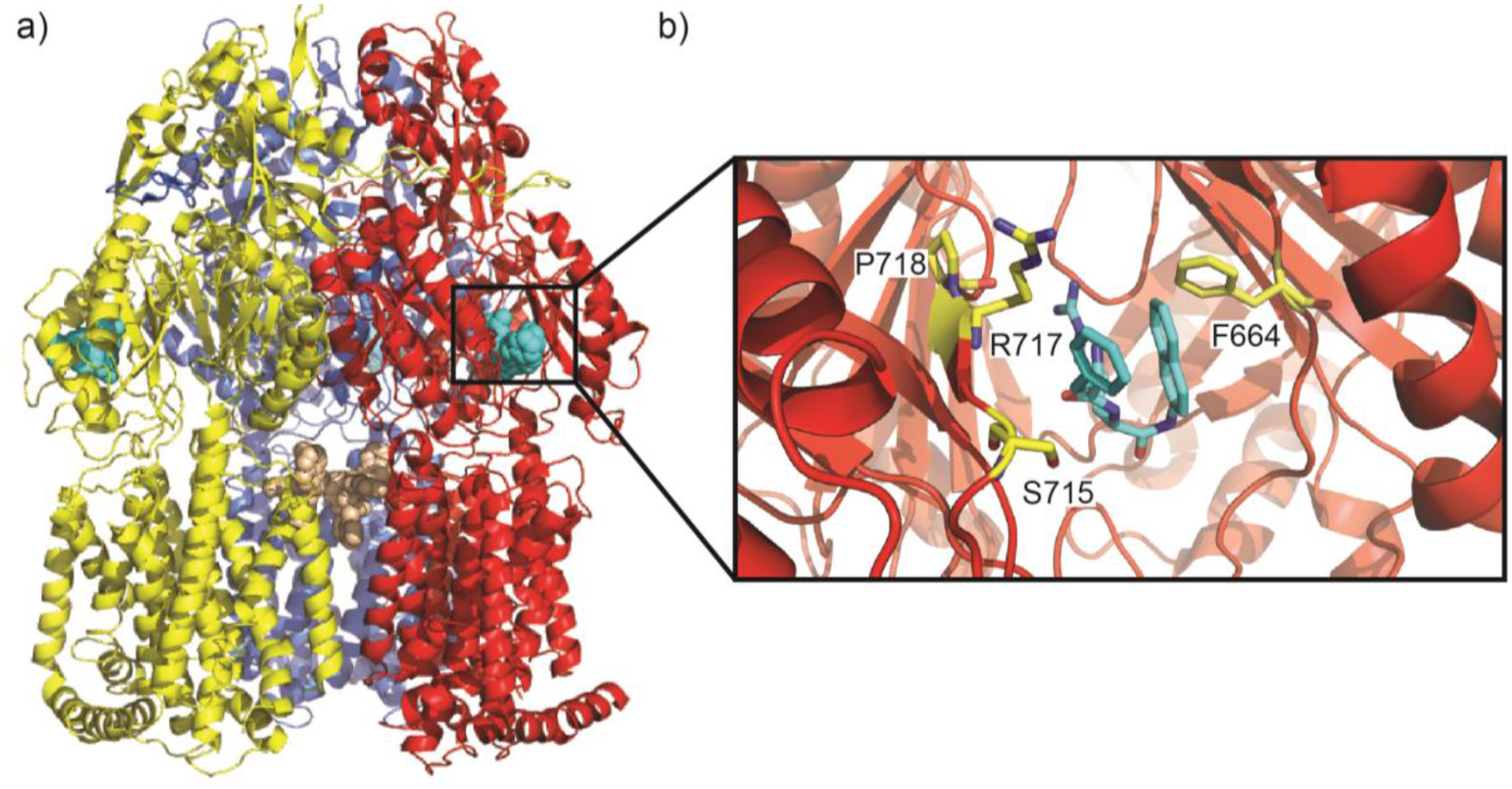
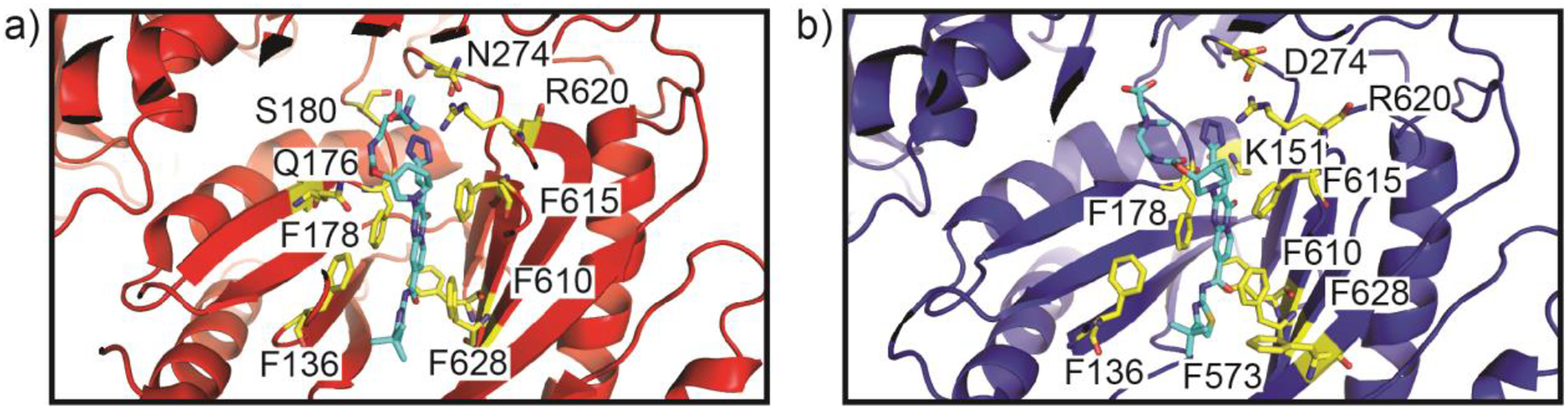
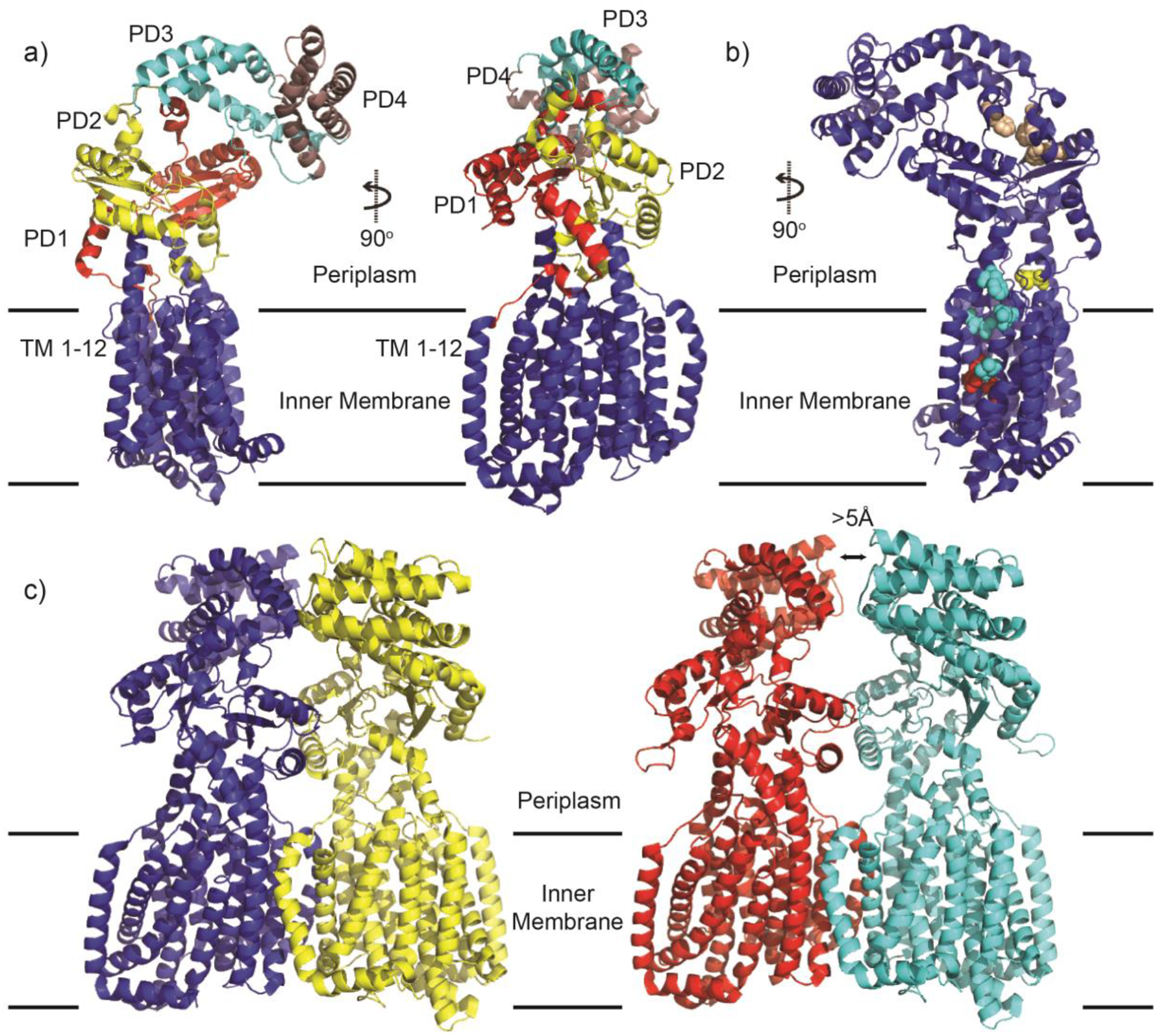
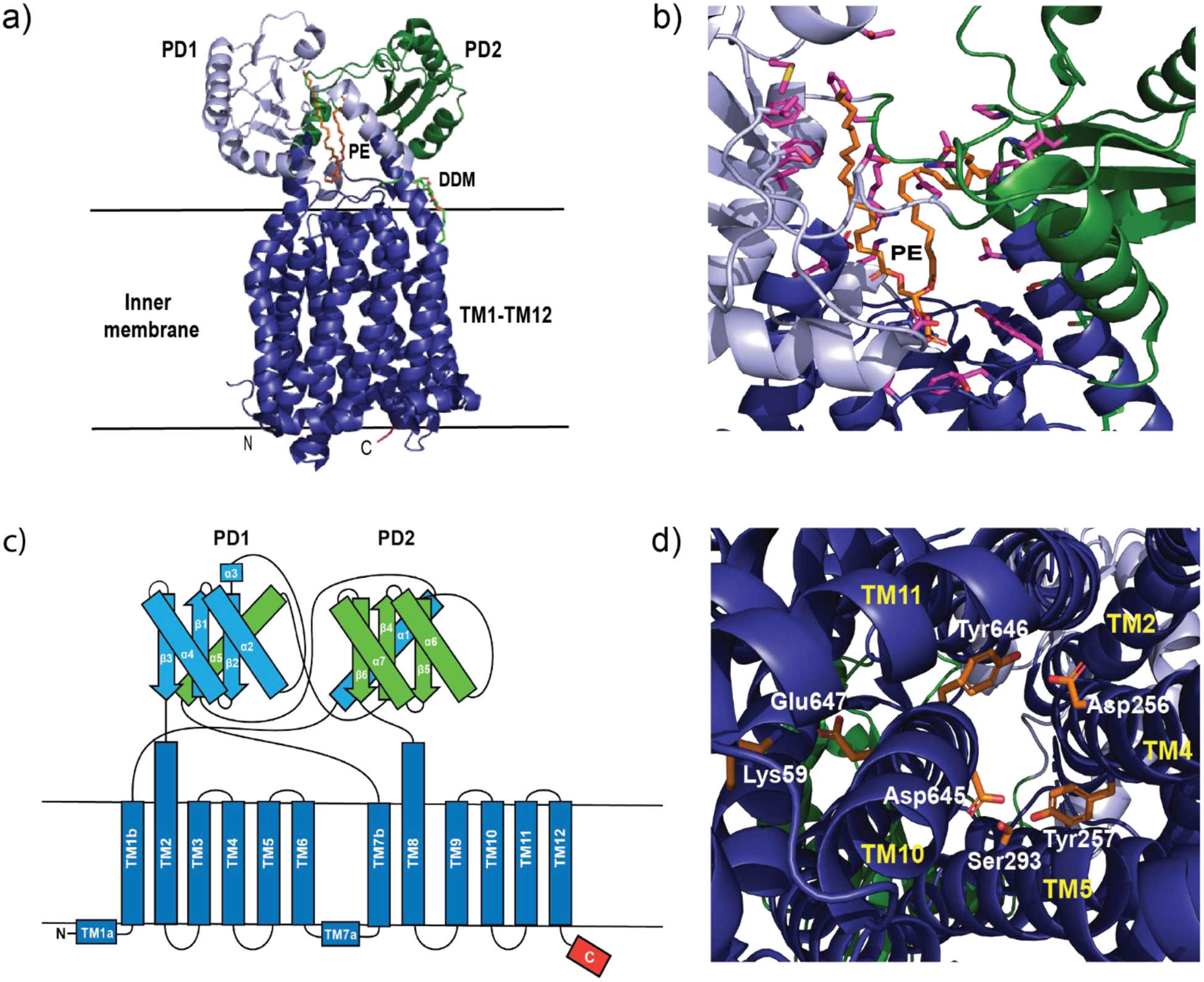
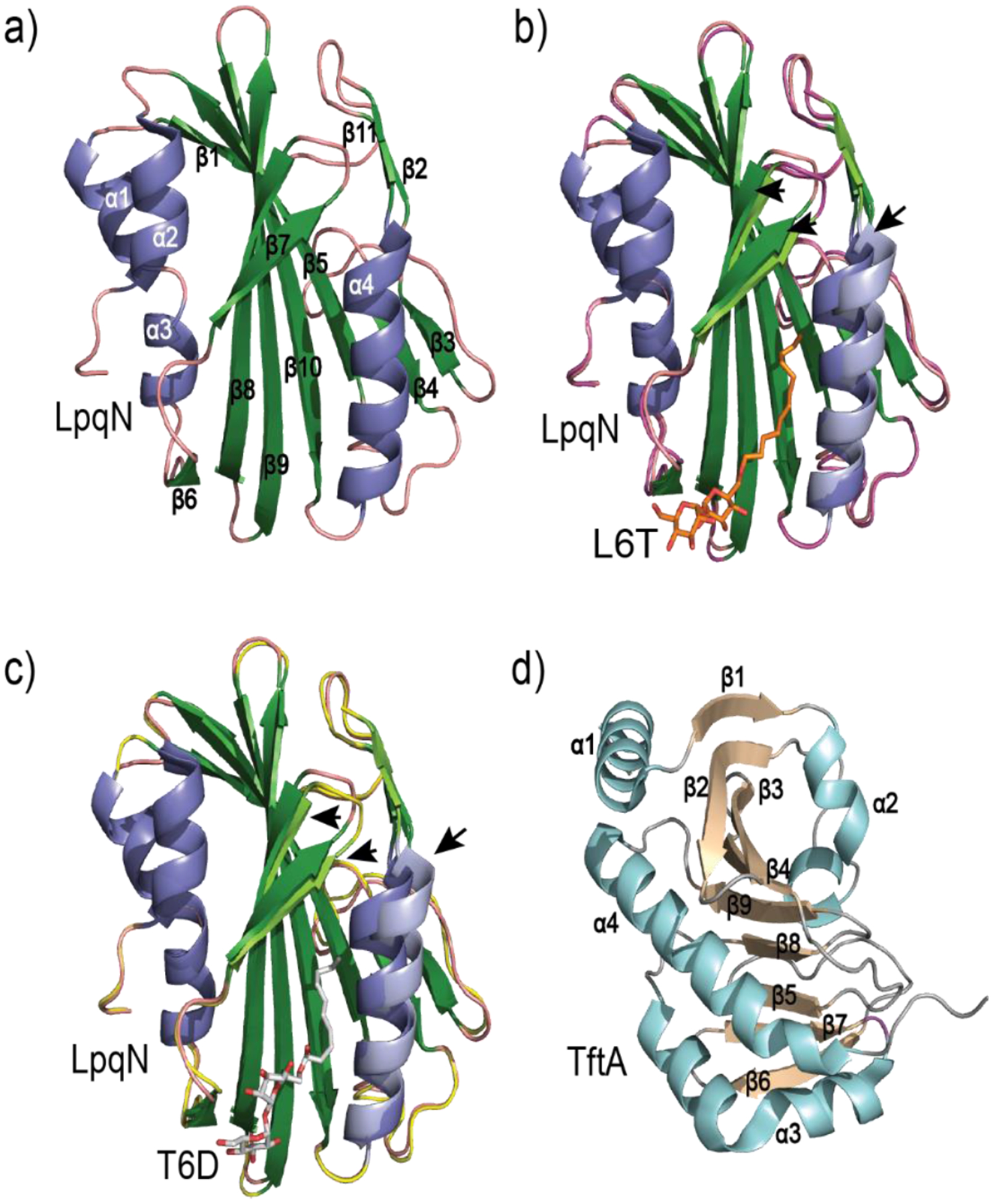
Similar articles
-
Multidrug Efflux Pumps and the Two-Faced Janus of Substrates and Inhibitors.Acc Chem Res. 2021 Feb 16;54(4):930-939. doi: 10.1021/acs.accounts.0c00843. Epub 2021 Feb 4. Acc Chem Res. 2021. PMID: 33539084 Free PMC article.
-
Structural and functional diversity of Resistance-Nodulation-Division (RND) efflux pump transporters with implications for antimicrobial resistance.Microbiol Mol Biol Rev. 2024 Sep 26;88(3):e0008923. doi: 10.1128/mmbr.00089-23. Epub 2024 Sep 5. Microbiol Mol Biol Rev. 2024. PMID: 39235227 Review.
-
Structural analysis of resistance-nodulation cell division transporters.Microbiol Mol Biol Rev. 2024 Jun 27;88(2):e0019823. doi: 10.1128/mmbr.00198-23. Epub 2024 Mar 29. Microbiol Mol Biol Rev. 2024. PMID: 38551344 Review.
-
Structures and Efflux Mechanisms of the AcrAB-TolC Pump.Subcell Biochem. 2024;104:1-16. doi: 10.1007/978-3-031-58843-3_1. Subcell Biochem. 2024. PMID: 38963480 Review.
-
Tripartite efflux pumps of the RND superfamily: what did we learn from computational studies?Microbiology (Reading). 2023 Mar;169(3):001307. doi: 10.1099/mic.0.001307. Microbiology (Reading). 2023. PMID: 36972322 Free PMC article. Review.
Cited by
-
Multidrug Efflux Pumps and the Two-Faced Janus of Substrates and Inhibitors.Acc Chem Res. 2021 Feb 16;54(4):930-939. doi: 10.1021/acs.accounts.0c00843. Epub 2021 Feb 4. Acc Chem Res. 2021. PMID: 33539084 Free PMC article.
-
A small molecule that disrupts S. Typhimurium membrane voltage without cell lysis reduces bacterial colonization of mice.PLoS Pathog. 2022 Jun 10;18(6):e1010606. doi: 10.1371/journal.ppat.1010606. eCollection 2022 Jun. PLoS Pathog. 2022. PMID: 35687608 Free PMC article.
-
Characterization and Molecular Determinants for β-Lactam Specificity of the Multidrug Efflux Pump AcrD from Salmonella typhimurium.Antibiotics (Basel). 2021 Dec 6;10(12):1494. doi: 10.3390/antibiotics10121494. Antibiotics (Basel). 2021. PMID: 34943706 Free PMC article.
-
Inhibition Mechanism of Anti-TB Drug SQ109: Allosteric Inhibition of TMM Translocation of Mycobacterium Tuberculosis MmpL3 Transporter.J Chem Inf Model. 2023 Aug 28;63(16):5356-5374. doi: 10.1021/acs.jcim.3c00616. Epub 2023 Aug 17. J Chem Inf Model. 2023. PMID: 37589273 Free PMC article.
-
Cryo-EM structure and resistance landscape of M. tuberculosis MmpL3: An emergent therapeutic target.Structure. 2021 Oct 7;29(10):1182-1191.e4. doi: 10.1016/j.str.2021.06.013. Epub 2021 Jul 8. Structure. 2021. PMID: 34242558 Free PMC article.
References
-
- WHO. https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance. Accessed May 4–6, 2020.
-
- Antibiotic Resistance Threats in the United States;2019. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-re... Accessed May 4–6, 2020.
-
- D’Costa VM; King CE; Kalan L; Morar M; Sung WWL; Schwarz C; Froese D; Zazula G; Calmels F; Debruyne R; Golding GB; Poinar HN; Wright GD Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. - PubMed
-
- Allen HK; Donato J; Wang HH; Cloud-Hansen KA; Davies J; Handelsman J Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol 2010, 8, 251–259. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
