

Genomic control of metastasis
- PMID: 33144692
- PMCID: PMC7782491
- DOI: 10.1038/s41416-020-01127-6
Genomic control of metastasis
Abstract
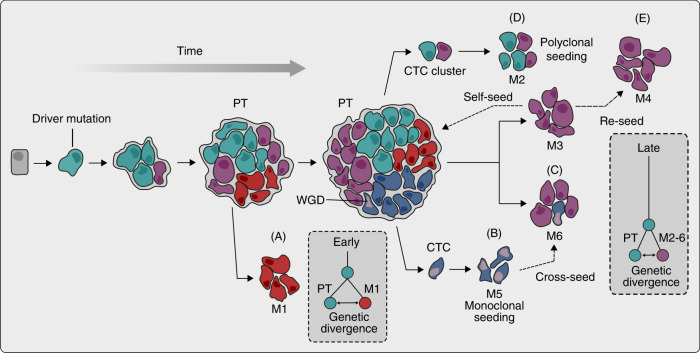
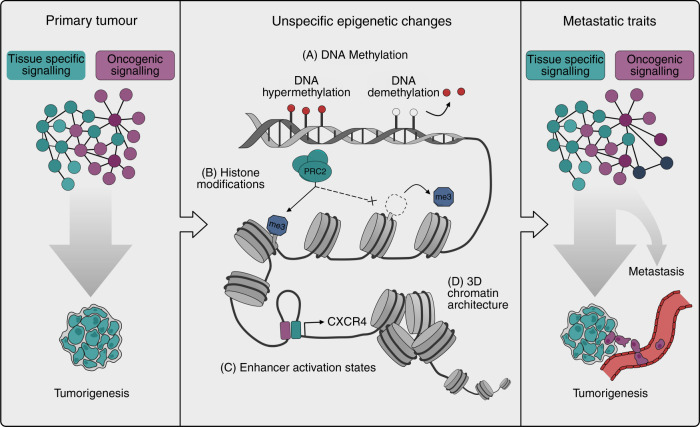
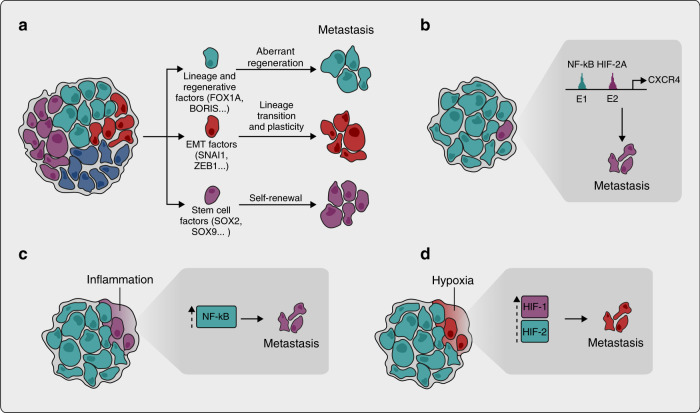
Metastasis remains the leading cause of cancer-associated mortality, and a detailed understanding of the metastatic process could suggest new therapeutic avenues. However, how metastatic phenotypes arise at the genomic level has remained a major open question in cancer biology. Comparative genetic studies of primary and metastatic cancers have revealed a complex picture of metastatic evolution with diverse temporal patterns and trajectories to dissemination. Whole-genome amplification is associated with metastatic cancer clones, but no metastasis-exclusive driver mutations have emerged. Instead, genetically activated oncogenic pathways that drive tumour initiation and early progression acquire metastatic traits by co-opting physiological programmes from stem cell, developmental and regenerative pathways. The functional consequences of oncogenic driver mutations therefore change via epigenetic mechanisms to promote metastasis. Increasing evidence is starting to uncover the molecular mechanisms that determine how specific oncogenic drivers interact with various physiological programmes, and what triggers their activation in support of metastasis. Detailed insight into the mechanisms that control metastasis is likely to reveal novel opportunities for intervention at different stages of metastatic progression.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Drivers of cancer metastasis - Arise early and remain present.Biochim Biophys Acta Rev Cancer. 2024 Mar;1879(2):189060. doi: 10.1016/j.bbcan.2023.189060. Epub 2023 Dec 25. Biochim Biophys Acta Rev Cancer. 2024. PMID: 38151195 Review.
-
Cancer, metastasis, and the epigenome.Mol Cancer. 2024 Aug 2;23(1):154. doi: 10.1186/s12943-024-02069-w. Mol Cancer. 2024. PMID: 39095874 Free PMC article. Review.
-
Epigenetic drivers of tumourigenesis and cancer metastasis.Semin Cancer Biol. 2018 Aug;51:149-159. doi: 10.1016/j.semcancer.2017.08.004. Epub 2017 Aug 12. Semin Cancer Biol. 2018. PMID: 28807546 Review.
-
Mutational drivers of cancer cell migration and invasion.Br J Cancer. 2021 Jan;124(1):102-114. doi: 10.1038/s41416-020-01149-0. Epub 2020 Nov 18. Br J Cancer. 2021. PMID: 33204027 Free PMC article. Review.
-
Cancer Genome Evolutionary Trajectories in Metastasis.Cancer Cell. 2020 Jan 13;37(1):8-19. doi: 10.1016/j.ccell.2019.12.004. Cancer Cell. 2020. PMID: 31935374 Review.
Cited by
-
A unique interplay of access and selection shapes peritoneal metastasis evolution in colorectal cancer.bioRxiv [Preprint]. 2024 Sep 27:2024.09.25.614736. doi: 10.1101/2024.09.25.614736. bioRxiv. 2024. PMID: 39386634 Free PMC article. Preprint.
-
Divergent iron regulatory states contribute to heterogeneity in breast cancer aggressiveness.iScience. 2024 Aug 3;27(9):110661. doi: 10.1016/j.isci.2024.110661. eCollection 2024 Sep 20. iScience. 2024. PMID: 39262774 Free PMC article.
-
Review of Current Principles of the Diagnosis and Management of Brain Metastases.Front Oncol. 2022 May 24;12:857622. doi: 10.3389/fonc.2022.857622. eCollection 2022. Front Oncol. 2022. PMID: 35686091 Free PMC article. Review.
-
Metastasis, an Example of Evolvability.Cancers (Basel). 2021 Jul 21;13(15):3653. doi: 10.3390/cancers13153653. Cancers (Basel). 2021. PMID: 34359555 Free PMC article.
-
Clinicopathologic and genomic characterizations of brain metastases using a comprehensive genomic panel.Front Med (Lausanne). 2022 Nov 24;9:947456. doi: 10.3389/fmed.2022.947456. eCollection 2022. Front Med (Lausanne). 2022. PMID: 36507516 Free PMC article.
References
-
- Fidler IJ. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. - PubMed
-
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. - PubMed
-
- Boire A, Brastianos PK, Garzia L, Valiente M. Brain metastasis. Nat. Rev. Cancer. 2020;20:4–11. - PubMed
-
- Amit M, Na’Ara S, Gil Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer. 2016;16:399–408. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
